






- Molecular Structure, Vibrational, Spectral Investigation and Quantum Chemical DFT Calculations of Poly(N-isopropyl acrylamide-co-nbutyl methacrylate)
Güzin Pihtili, Seda Hekim*,†
 , and Mustafa Ersin Pekdemir**
, and Mustafa Ersin Pekdemir**Department of Medical Services and Techniques, Munzur University, Tunceli, 62000, Turkey *Department of Physics, Faculty of Science, Firat University, Elazig, 23119, Turkey **Department of Chemistry, Faculty of Science, Firat University, Elazig, 23119, Turkey
- Poly(N-isopropyl acrylamide-co-nbutyl methacrylate)의 분자구조, 진동, 스펙트럼 조사 및 양자화학 DFT 계산
Reproduction, stored in a retrieval system, or transmitted in any form of any part of this publication is permitted only by written permission from the Polymer Society of Korea.
A new copolymer poly(N-isopropylacrylamide-co-nbutylmethacrylate) was synthesized by free radical solution polymerization process using 2,2′-Azobis(2-methylpropionitrile) as an initiator in 1,4-dioxane at 70 °C. The structure of the copolymer was characterized by Fourier Transform Infrared Spectrometer (FTIR), the proton nuclear magnetic resonance (1H NMR), and the ultraviolet-visible spectroscopy (UV-vis) method. Theoretically, density functional theory (DFT) (B3LYP) was utilized to identify the molecular geometry and then examine the vibrational frequencies and electrical characteristics of this molecule, with the 6-31G basis set in the ground state. In dichloromethane solution, UV-vis spectra of the molecule were calculated using the Time-Dependent DFT method. 1H NMR chemical shifts have been calculated with GIAO approximation, and the calculated results show good agreement with experimental chemical shifts.
Functional poly N-isopropylacryl-amide (PNIPAMs) are polymers with many applications. Nanotechnology, drug delivery systems, purification, separation, and bio-nervous system are some of the usage areas. The structural and spectroscopic properties of the P(NIPAM-co-BtMA) molecule were determined utilizing FTIR, UV-Vis, and 1H NMR experimental methods. The calculated molecular structure, vibrational frequencies, and chemical shift values with the B3LYP/6-31G level differ slightly from experimental measurements as a result. Despite these differences, the computed results showed that the theoretical approximation was accurate.
Keywords: N-isopropylacrylamide, copolymer, HOMO and LUMO, density functional theory, infrared spectrometer.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The development of macromolecular chemistry has made functional polymers more attractive both commercially and scientifically. Functional polymers, which can be obtained by polymerization or copolymerization of monomers with desired functional chains, can also be synthesized by chemical modification.1 Amide group-containing polymers are macromolecules with good mechanical-thermal properties and high-temperature process ability.2 Functional poly N-isopropylacrylamide (PNIPAMs) are polymers with a wide range of applications.3 Functional PNIPAMs have been synthesized and employed in many fields. Nanotechnology, drug delivery systems, purification, separation, and bio-nervous system are some of the usage areas.4-10 Free radical polymerization (FRP) is a well-known method, and product design provides from knowing the relative ratios of polymer reactions, as well as, more importantly, the physics that governs these rates. FRP continues over a chain mechanism. Polymerization consists of four different types of reactions involving free radicals. FRP is inhibited by the presence of oxygen or delayed from entering the polymerization environment. Conventional radical polymerization is applied as bulk, solution, suspension, dispersion, precipitation, and microemulsion.10 Charge transfer complexes can be initiators of radical polymerization. These complexes are structures in the property of an electron donor-electron acceptor in which a partial electronic charge transfer takes place from the donor to the acceptor molecule.11-14
Quantum mechanical laws began to be applied to atoms and molecules with the development of quantum theory. Quantum chemical calculations are computed using molecular modeling programs to supplement experimental research or forecast outcomes that can be reached without the need for experiments. Theoretical computation approaches and associated fields of study have exploded in prominence in recent years, and they now serve a crucial role in determining a wide range of molecular behaviors. When performing computational quantum chemistry operations that include the ab initio calculation of the electronic structures of many particle systems, the density functional theory (DFT) is widely used. Furthermore, it is clear from the literature that the DFT approach is used to accurately calculate all of the molecular geometry, dipole moment, and spectroscopic data, including NMR and NMR chemical shifts, IR, and UV/Vis of organic and inorganic compounds.15-22
This work is concerned with modeling the propagation and chain transfer reactions in free radical polymerization of N-isopropylacrylamide (NIPAM) and n-butylmethacrylate(BtMA) and in the study, firstly the P(NIPAM-co-BtMA) molecule was synthesized, and then the spectroscopic and electronic characteristics of P(NIPAM-co-BtMA) were examined both experimentally and theoretically. Theoretical calculations of the investigated compound were made using the Gaussian 09W23 program and the DFT method. In the DFT calculations, the B3LYP mixed functional was used, which is one of the most widely used exchange-correlation functional and includes Becke's three-parameter exchange functional and Lee, Yang, and Parr's correlation functional.24,25 All these calculations were done using 6-31G basis set. The spectroscopic behaviors, which find theoretically, were compared with some experimental data and it has been determined that the experimental data and the theoretical findings are very well congruent.
Materials. Synthesis of poly(N-isopropylacrylamide-co-n-butylmethacrylate) was achieved at the laboratory. NIPAM and n-BtMA monomers were purchased and used as received by Sigma-Aldrich, an American company. 2,2'-Azobis(2-methylpropionitrile) (AIBN) (Sigma-Aldrich, 98%) was used as free radical initiator. All of the other chemicals used were of analytical purity and used as received commercially.
Characterization Experiments. The characterization analyzes contained Fourier Transform Infrared (FTIR) and nuclear magnetic resonance (NMR) spectroscopy. An FTIR spectroscopic way was utilized to get structural information on poly(NIPAM-co-BtMA). It was recorded on a PerkinElmer device. 1H NMR spectrometer (Bruker AVANCE III, 400 MHz) was used to obtain NMR-hydrogen spectroscopy particulars of the copolymer.
Synthesis of the Poly(NIPAM-co-BtMA). The poly(N-isopropylacrylamide-co-n-butylmethacrylate) was prepared by free-radical polymerization in 1,4 dioxane. The synthesis of the copolymer precursor was conducted at 50% w/w monomers. In brief, NIPAM (1 g), n-BtMA (1 g), and 1,4 dioxane (20 mL) were added in a polymerization tube followed by magnetic stirring. The free-radical polymerization initiator AIBN was added at 1% of the total monomer mass ratio. Argon gas was passed through the solution in the tube for 30 min, and the polymer reaction was continued in the oil bath for 8 h (Figure 1). The copolymer was precipitated into n-hexane and put in to under vacuum for drying.
Theoretical Calculation Method. Computational approaches have been increasingly popular in recent years for elucidating and predicting molecular behavior. Molecule behavior can thus be anticipated without the need for experimentation. In this case, information on numerous qualities that cannot be obtained empirically and corroborate experimental data is provided by applying DFT, one of the theoretical calculations. All calculations were performed using the GAUSSIAN-09W23 software package and GaussView, Rev 5.0.826 molecular visualization programs. In the DFT calculations, the B3LYP mixed functional, which is one of the most widely used exchange-correlation functional, includes Becke's three-parameter exchange functional27 and Lee, Yang, and Parr's correlation functional.28 The geometric optimization of the molecule, its geometric parameters, and energy values was derived using the DFT method with the base set 6-31G. Using the same approach and basis set, NMR, UV, and IR spectra were theoretically computed from optimized geometry. The NMR chemical shift values of the compounds were determined using the gauge-independent atomic orbital (GIAO) method,29,30 with TMS [tetramethylsilane, Si(CH3)4] as a reference. This molecule's theoretically available spectroscopic data were compared to experimental data.

|
Figure 1 Synthesis of poly(NIPAM-co-BtMA). |
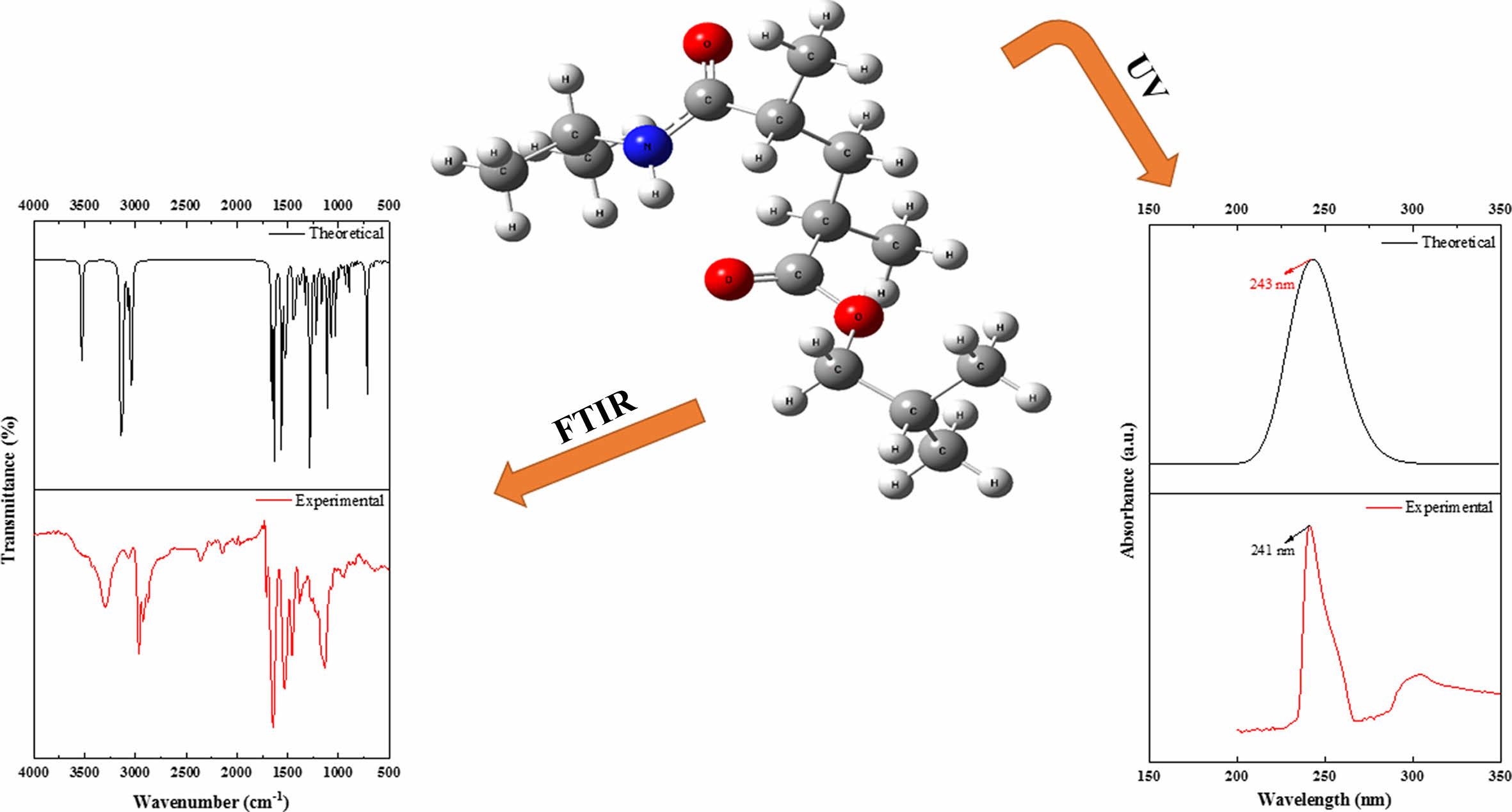
Molecular Structure. Changes in the energy and other properties of molecules are caused by structural and positional changes. The molecule's approximate most stable structure is obtained by geometric optimization. As a result, a stable, low-energy molecule structure is found. Figure 2 shows the theoretical geometric structure of poly(NIPAM-co-BtMA), which was determined by doing the necessary calculations for just one unit.
HOMO-LUMO Analysis and Electronic Properties. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies are the most well-known quantum chemical parameters. These orbitals, which are also referred to as frontier molecular orbitals, are crucial in determining both the electronic and optical properties of a molecule, particularly in chemical reactions. Figure 3 depicts the HOMO and LUMO surfaces and energies of the investigated structure. The HOMO energy is a measure of a molecule's ability to donate electrons and is related to its ionization potential. Moreover, the ability of a molecule to gain electrons is represented by LUMO energy, which is directly connected to electron affinity. The energy difference between HOMO and LUMO can be used to calculate a molecule's chemical stability. As can be seen from the figure, the energy difference between HOMO and LUMO is 5.684 eV. The fact that this energy range is wide implies that the molecule is in a stable state. The HOMO and LUMO energies were also used to compute electronegativity, chemical potential, chemical hardness, and chemical softness. Table 1 lists these molecules electrical characteristics. The molecule is extremely stable, with negligible chemical reactivity and great kinetic stability, as shown in Table 1.
Vibrational Frequencies. The vibrational energy levels associated with chemical bonds in a sample are measured using FTIR spectroscopy, which is one of the spectroscopic methods. The peaks in the FTIR spectra correspond to specific vibrations of chemical bonds or functional groups inside molecules.31 The experimental and theoretical structure of the poly(NIPAM-co-BtMA) copolymer system was determined by FTIR spectroscopy in the region of 4000-400 cm-1 and was shown in Figure 4. The copolymer system has characteristic absorption peaks; the band at 3300 cm-1 are determined to the stretch for the hydrogen-bonded N-H group, is strong peak at 1650 cm-1 assigned to (C=O) a stretching vibration, and at 1540 cm-1 corresponds to N-H vibration of the amide group (-CONH) in NIPA. In the nBtMA; Aliphatic C-H stretching vibration at 2950-2862 cm-1, methacrylate OC= O ester carbonyl at 1728 cm-1, aliphatic C-H bending vibration at 1455-1367 cm-1, respectively. Theoretically, the stretch for the hydrogen-bonded N-H group can be observed at 3526 cm-1, (C=O) a stretching vibration can be observed at 1691 cm-1. The aliphatic C-H stretching vibration appears at 3123-3044 cm-1, the methacrylate -O-C= O ester carbonyl at 1667 cm-1, the aliphatic C-H bending vibration at 1469-1381 cm-1, respectively. The results of calculated vibrational frequencies and experimental data were given in Table 2. It can be observed that the experimental and theoretical results are in agreement. However, utilizing a single unit in computations and viewing electron correlations as partial helps explain the discrepancies between theoretical and actual data (particularly N-H stretching vibration and C-H aliphatic stretching states).
NMR Analysis. 1H NMR is a significant way used to research the structure of materials, characterize and analyze solid samples,32 and is used to investigate all compounds.33 Amine and amide-containing compounds are among the best examples showing the effect of the solvent on N-H hydrogen NMR chemical shifts.34,35 In our study, experimental and theoretical NMR data were compared. Functional groups of the copolymer were obtained for 1H NMR chemical shift calculations. The NMR chemical shift calculations in these molecular structures were done theoretically using the Gauge-Including Atomic Orbitals NMR attitude and the 6-31G base set in the DFT approach. Table 3 shows the theoretical and experimentally determined NMR chemical shifts of poly (NIPAM-co-BtMA). The experimental 1H NMR spectrum of copolymer showed the significate bans at δ; -NHC=O in NIPA units were observed at 6.50-7.50 ppm, at 3.8 ppm for protons next to the amide in NIPAM, CH2- bound to ester carbonyl in n-BMA units at 3.9 ppm protons signal, at 0.6-2.1 ppm for -CH2 and -CH3 protons in the polymer chain. When the theoretical and experimental chemical shift results in Table 3 are compared, the experimental data and the findings of the theoretical calculations are found to be in good agreement. The poly(NIPAM-co-BtMA) compound's theoretical and experimental 1H chemical shift values are compatible, indicating that the theoretically discovered molecular structure is accurate. Because the correct theoretical calculation of chemical shift values is contingent on the molecular structure being correctly determined.
UV-Vis Absorption Analysis. UV-vis spectroscopy of poly (NIPAM-co-BtMA) was investigated both experimentally and theoretically. Experimental UV-vis spectrum of poly (NIPAM-co-BtMA) was recorded in the range of 150 and 350 nm in dichloromethane solution with PerkinElmer Lambda 45 UV/VIS spectrometer. In terms of theoretical calculations, time-dependent density functional theory (TD-DFT) is the most common method for predicting electrically excited states and explaining UV-Vis spectra. In this case, the TD-DFT approach was used to calculate oscillator strengths, vertical excitation energy, and wavelengths of our calculated results of the title molecule. A solvent effect is particularly significant in analyzing UV-vis spectrum data because the solvents might link together in hydrogen bonds, leading to strong inter- or intramolecular interactions. Theoretical calculations for UV-vis were performed using TD-DFT/B3LYP with 6-31G basis set in dichloromethane solution. Both experimental and theoretical computed UV-vis spectra of poly (NIPAM-co-BtMA) are shown in Figure 5. The strong absorption maxima for poly (NIPAM-co-BtMA) were determined at 241nm and 243nm experimentally and theoretically, respectively, as shown in Figure 5.

|
Figure 2 The theoretical optimized molecular structure. |

|
Figure 3 Three-dimensional representation of HOMO and LUMO molecular orbitals of molecule and their energy values. |

|
Figure 4 Experimental and theoretical FTIR spectra of poly (NIPAM-co-BtMA). |

|
Figure 5 Experimental and theoretical UV-vis spectra of poly(NIPAMco-nBtMA). |

|
Table 2 The Calculated and Experimental Vibrational Frequencies of the Cop olymer |

|
Table 3 The Experimental and Calculated (GIAO) 1 H NMR Chemical Shifts (concerning TMS, all values in ppm) of Poly (NIPAM-co-BtMA) |

The structural and spectroscopic properties of the P(NIPAM-co-BtMA) molecule were determined utilizing FTIR, UV-Vis, and 1H NMR experimental methods in this study. The molecular structure, vibrational frequencies, and chemical shift values were computed with the B3LYP/6-31G level and compared to the experimental data to back up the findings. As a result, the theoretical and experimental values are found to have good coherence. 1H NMR spectra were obtained, and the results were compared to experimental data. Using the TD-DFT approximation, electronic absorption spectra were determined. The calculations were done using an isolated single molecule in the gas phase. Experimental data, on the other hand, is derived from the solid state of matter, and these measurements include intermolecular interactions. Theoretical calculations and experimental measurements differ slightly as a result. Despite these differences, the computed results showed that the theoretical approximation was accurate.
- 1. Coskun, M.; Temuz, M. M.; Koca, M. Thermal Degradation Behaviour of Poly[(2-hydroxy-3-phenoxy) propyl methacrylate] and Poly[(2-hydroxy-3-tetrahydrofurfuryloxy) propyl methacrylate]. Polym. Degrad. Stab. 2003, 81, 95-102.
-
- 2. Biryan, F.; Pihtili, G. Fabrication of a Novel Acrylate Polymer Bearing Chalcone and Amide Groups and Investigation of its Thermal and Isoconversional Kinetic Analysis. J. Therm. Anal. Calorim. 2020, 139, 3857-3870.
-
- 3. İlboğa, S.; Pekdemir, E.; Coşkun, M. Cloud Point Temperature, Thermal and Dielectrical Behaviors of Thermosensitive Block Copolymers Based N-Isopropylacrylamide. Polymer Science, Series B 2019, 61, 32-41.
-
- 4. Qiu, N.; Li, Y.; Han, S.; Cui, G.; Satoh, T.; Kakuchi, T. et al. Synthesis of Star Poly(N-isopropylacrylamide) with End-group of Zinc-porphyrin via ATRP and Its Photocatalytic Activity Under Visible Light. J. Photochem. Photobiol. A: Chem. 2014, 283, 38-44.
-
- 5. Ankareddi, I.; Brazel, C. S. Synthesis and Characterization of Grafted Thermosensitive Hydrogels for Heating Activated Controlled Release. Int. J. Pharm. 2007, 336, 241-247.
-
- 6. Fujimoto, K.; Iwasaki, C.; Arai, C.; Kuwako, M.; Yasugi, E. Control of Cell Death by the Smart Polymeric Vehicle. Biomacromolecules 2000, 1, 515-518.
-
- 7. Zhang, X.-Z.; Zhuo, R.-X.; Cui, J.-Z.; Zhang, J.-T. A Novel Thermo-responsive Drug Delivery System with Positive Controlled Release. Int. J. Pharm. 2002, 235, 43-50.
-
- 8. Matsuda, T.; Saito, Y.; Shoda, K. Cell Sorting Technique Based on Thermoresponsive Differential Cell Adhesiveness. Biomacromolecules 2007, 8, 2345-2349.
-
- 9. Pennadam, S. S.; Lavigne, M. D.; Dutta, C. F.; Firman, K.; Mernagh, D.; Górecki, D. C. et al. Control of a Multisubunit DNA Motor by a Thermoresponsive Polymer Switch. J. Am. Chem. Soc. 2004, 126, 13208-13209.
-
- 10. Lu, X.; Zhang, L.; Meng, L.; Liu, Y. Synthesis of Poly(N-isopropylacrylamide) by ATRP Using a Fluorescein-based Initiator. Polym. Bull. 2007, 59, 195-206.
-
- 11. Matyjaszewski, K.; Davis, T. P. Handbook of Radical Polymerization; A John Wiley & Sons. Inc.: Hoboken, 2002.
-
- 12. Gencoglu, T.; Eren, T. N.; Lalevee, J.; Avci, D. Photoinitiating Systems Based on Poly(ethylene imine) for Michael Addition and Free Radical Photopolymerization. J. Photochem. Photobiol. A: Chem. 2021, 404, 112959.
-
- 13. Wang, D.; Garra, P.; Lakhdar, S.; Graff, B.; Fouassier, J. P.; Mokbel, H. et al. Charge Transfer Complexes as Dual Thermal and Photochemical Polymerization Initiators for 3D Printing and Composites Synthesis. ACS Appl. Polym. Mater. 2019, 1, 561-570.
-
- 14. Garra, P.; Graff, B.; Morlet-Savary, F.; Dietlin, C.; Becht, J.-M.; Fouassier, J.-P. et al. Charge Transfer Complexes as Pan-scaled Photoinitiating Systems: from 50 μm 3D Printed Polymers at 405 nm to Extremely Deep Photopolymerization (31 cm). Macromol. 2018, 51, 57-70.
-
- 15. Karakurt, T.; Cukurovali, A.; Subasi, N. T.; Onaran, A.; Ece, A.; Eker, S. et al. Experimental and Theoretical Studies on Tautomeric Structures of a Newly Synthesized 2,2′-(hydrazine-1, 2-diylidenebis (propan-1-yl-1-ylidene)) Diphenol. Chem. Phys. Lett. 2018, 693, 132-145.
-
- 16. Karakurt, T.; Cukurovali, A.; Subasi, N. T.; Kani, I. Molecular Structure and Computational Studies on 2-((2-(4-(3-(2,5-dimethylphenyl)-3-methylcyclobutyl) thiazol-2-yl) hydrazono) methyl) Phenol Monomer and Dimer by DFT Calculations. J. Mol. Struct. 2016, 1125, 433-442.
-
- 17. Barim, E.; Akman, F. Synthesis, Characterization and Spectroscopic Investigation of N-(2-acetylbenzofuran-3-yl) Acrylamide Monomer: Molecular Structure, HOMO-LUMO Study, TD-DFT and MEP Analysis. J. Mol. Struct. 2019, 1195, 506-513.
-
- 18. Demir, P.; Akman, F. Molecular Structure, Spectroscopic Characterization, HOMO and LUMO Analysis of PU and PCL Grafted onto PEMA-co-PHEMA with DFT Quantum Chemical Calculations. J. Mol. Struct. 2017, 1134, 404-415.
-
- 19. Evecen, M.; Tanak, H. Quantum Chemical Studies on the Molecular Structure, Spectroscopic and Electronic Properties of (6-Methoxy-2-oxo-2H-chromen-4-yl)-methyl Pyrrolidine-1-carbodithioate. Materials Science-Poland 2016, 34, 886-904.
-
- 20. Evecen, M.; Tanak, H.; Tinmaz, F.; Dege, N.; İlhan, İ. Ö. Experimental (XRD, IR and NMR) and Theoretical Investigations on 1-(2-nitrobenzoyl) 3,5-bis(4-methoxyphenyl)-4, 5-dihydro-1H-pyrazole. J. Mol. Struct. 2016, 1126, 117-126.
-
- 21. Shahab, S.; Sheikhi, M.; Filippovich, L.; Ignatovich, Z.; Muravsky, A.; Alnajjar, R. et al. Optimization, Spectroscopic (FTIR, Excited States, UV/Vis) Studies, FMO, ELF, LOL, QTAIM and NBO Analyses and Electronic Properties of Two New Pyrimidine Derivatives. Chin. J. Struct. Chem. 2019, 38, 1615-1639.
-
- 22. Shahab, S.; Sheikhi, M.; Filippovich, L.; Khaleghian, M.; Dikusar, E.; Yahyaei, H. et al. Spectroscopic Studies (geometry Optimization, E → Z Isomerization, UV/Vis, Excited States, FTIR, HOMO-LUMO, FMO, MEP, NBO, Polarization) and Anisotropy of Thermal and Electrical Conductivity of New Azomethine Dyes in Stretched Polymer Matrix. Silicon. 2018, 10, 2361-2385.
-
- 23. Frisch, M.; Trucks, G.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R. et al. Gaussian 09, Revision d. 01; Gaussian. Inc.: Wallingford, 2009; p 201.
- 24. Hohenberg, P.; Kohn, W. Density Functional Theory (DFT). Phys. Rev. 1964, 136, B864.
- 25. Jones, R. O.; Gunnarsson, O. The Density Functional Formalism, Its Applications and Prospects. Rev. Mod. Phys. 1989, 61, 689.
-
- 26. Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, 2009.
- 27. Becke, A. D. Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098.
-
- 28. Becke, A. D. A New Mixing of Hartree-Fock and Local Density‐functional Theories. J. Chem. Phys. 1993, 98, 1372-1377.
-
- 29. Merrick, J. P.; Moran, D.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683-11700.
-
- 30. Dodds, J. L.; McWeeny, R.; Sadlej, A. J. Self-consistent Perturbation Theory: Generalization for Perturbation-dependent Non-orthogonal Basis Set. Mol. Phys. 1977, 34, 1779-1791.
-
- 31. Acemi A. Polymerization degree of chitosan affects structural and compositional changes in the cell walls, membrane lipids, and proteins in the leaves of Ipomoea purpurea: An FTIR spectroscopy study. Int. J. Biol. Macromol. 2020, 162, 715-722.
-
- 32. Yan, J.; Nie, W.; Zhang, H.; Xiu, Z.; Bao, Q.; Wang, H. et al. Synthesis and Performance Measurement of a Modified Polymer Dust Suppressant. Adv. Powder Technol. 2020, 31, 792-803.
-
- 33. Acikses, A.; Hekim, S.; Oksuz, F. Spectroscopic and Electronic Properties of a Copolymer and Its Metal Complexes: A Theoretical and Experimental Study. Chem. Phys. 2019, 527, 110469.
-
- 34. Da Silva, H. C.; De Almeida, W. B. Theoretical Calculations of 1H NMR Chemical Shifts for Nitrogenated Compounds in Chloroform Solution. Chem. Phys. 2020, 528, 110479.
-
- 35. Abraham, R. J.; Byrne, J. J.; Griffiths, L.; Perez, M. 1H Chemical Shifts in NMR: Part 23, the Effect of Dimethyl Sulphoxide versus Chloroform Solvent on 1H Chemical Shifts. Magn. Reson. Chem. 2006, 44, 491-509.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2022; 46(5): 559-565
Published online Sep 25, 2022
- 10.7317/pk.2022.46.5.559
- Received on Apr 5, 2022
- Revised on Jul 18, 2022
- Accepted on Jul 20, 2022
Services
- Full Text PDF
- Abstract
- ToC
- Conflict of Interest
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Güzin Pihtili, Seda Hekim
-
Department of Medical Services and Techniques, Munzur University, Tunceli, 62000, Turkey
- E-mail: ssurucu@firat.edu.tr
- ORCID:
0000-0003-1932-6978
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr