






- Different Biopolymers' Effects on the Evaluation and Characterization of Floating Tablets Prepared by Lyophilization Technique to Improve the Quality Control Parameters
Rukiye Sevinç Özakar
 and Emrah Özakar†
and Emrah Özakar† Department of Pharmaceutical Technology, Faculty of Pharmacy, Atatürk University, Erzurum, Turkey
- 품질관리 향상을 위한 다양한 생체고분자의 영향과 동결건조 부유약제의 특성 분석
Reproduction, stored in a retrieval system, or transmitted in any form of any part of this publication is permitted only by written permission from the Polymer Society of Korea.
Conventional oral drug systems are unable to keep drug concentration within the therapeutic range, and administration of the dosage form several times a day can cause significant fluctuations in plasma drug concentration. Therefore, floating drug delivery systems are being developed. In this study, floating tablets with a model drug (ampicillin sodium-Amp.Na) were successfully prepared using different polymers by the lyophilization technique. This study aims to prepare and characterize floating lyophilized tablets to understand the effect of different biopolymers on quality control parameters more clearly. Within the scope of characterization studies, many parameters were evaluated successfully. All tablets showed 24-hour release and had a mesoporous structure, as understood from the Brunauer-Emmett-Teller (BET) analysis. It has been determined from the Fourier transform infrared (FTIR) and X-ray diffraction (XRD) analyses that Amp.Na doesn’t interact with excipients and is an amorphous form. These formulations can be used for other drugs in the future, and optimum properties can be easily characterized.
In this study, floating tablets with a model drug (ampicillin sodium-Amp.Na) were prepared using different polymers by the lyophilization technique successfully. All tablets showed 24-hour release and had a mesoporous structure, as understood from the Brunauer-Emmett-Teller (BET) analysis. It has been determined from the Fourier transform infrared (FTIR) and X-ray diffraction (XRD) analyzes that Amp.Na doesn¡¯t interact with excipients.
Keywords: floating tablet, lyophilization, polymers, release kinetics, Brunauer-Emmett-Teller, Fourier transform infrared, X-ray diffraction.
The authors would like to gratefully acknowledge the kind support of East Anatolian High Technology Research and Application Center (DAYTAM) of Atatürk University for FTIR, BET and XRD analyzes. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors thank financial support for this work from the Atatürk University Scientific Research Project Foundation (Grant Number: THD-2020-8291).
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Oral dosage forms have been an indispensable way of drug administration for humankind for centuries. Oral dosage forms are very diverse and have many advantages over other dosage forms. Oral drug administration is the easiest and most preferred way of administering therapeutic agents with a single dose to achieve the systemic effect since it can be used without any assistance in terms of patient compliance. The oral route has variable and multifaceted physiological conditions. This route allows the development of oral formulations that can selectively release the drug for optimum therapeutic benefit. Conventional drug systems are unable to keep drug concentration within the therapeutic range, and administration of the dosage form several times a day can cause significant fluctuations in plasma drug concentration. Therefore, new carriers are being developed for the oral delivery of drugs.1,2 Many drug delivery systems (e.g., solid dispersions, nano-sized drugs, or controlled release systems) have been investigated to increase the bioavailability or efficacy of oral dosage forms. Oral sustained release delivery systems can carry active ingredients in the human body that have both a short half-life in plasma and good absorption and thereby providing a longer duration of the therapeutic effect. However, these systems could not achieve sufficient efficacy for some active ingredients that act locally in the stomach, are absorbed in the proximal part of the gastrointestinal system, and have low solubility and stability in the gastrointestinal system.3-5 There are also disadvantages such as the short residence time of the dosage forms in the gastrointestinal tract, degradation of active substances due to the high activity of the gastrointestinal content, and unpredictable gastric emptying and motility. Gastric emptying is a complex process and makes the performance of drug delivery systems uncertain in “in vivo” studies. Gastroretentive drug delivery systems are useful systems that can prevent these uncertainties by increasing the duration of drug delivery systems in the stomach. The main rationale for developing gastroretentive controlled release systems is to maintain drug concentration in the absorption window, reduce the number of administrations and side effects and increase the effectiveness of drugs.6-8 The gastroretentive drug delivery systems provide more extended drug release and the residence time of the drugs in the gastrointestinal system until all the active ingredients are completely released for many hours. These systems have many advantages like a prolonged gastric emptying process, increased absorption, bioavailability and therapeutic efficiency, reduced drug wastes, improved solubility, and patient compliance. Also, it provides to deliver the drugs to the stomach and proximal small intestine locally.3,9 The preparation of gastroretentive drug delivery systems has been designed by different approaches like floating (low density), sinking (high density), swelling (expandable), mucoadhesive, and magnetic systems that proper gastric retention time and control the release rate of the drug.3-5,10The most popular and favorable of these systems are floating drug delivery systems because of not affected by the motility and content of the gastrointestinal tract.9 Floating drug delivery systems, also known as hydrodynamically controllable systems, are classified as effervescent and non-effervescent systems. These systems are low-density systems that have a hydrophilic character and can remain floating without affecting the gastric emptying rate in the stomach for a long time. With the gelation of the outer surface, a barrier is formed, and the release of the active substance can be controlled from the swelling matrix.11-13 After the finishing of the release of the active substance, its residues are removed along with the stomach contents. It is possible that these systems can control fluctuations in the drug plasma level, increase absorption, provide local effects in the stomach (such as antacids, antibiotics), decrease mucosal irritation/toxicity, and be produced easily.6,12,14 These systems are matrix-type systems prepared with the help of gel-forming or swellable hydrocolloid polymers (hydroxypropyl methylcellulose (HPMC), methylcellulose, sodium carboxymethylcellulose (NaCMC), agar, polyacrylate, polycarbonate, polystyrene, carbopol, sodium alginate (SA) or chitosan) and/or various effervescent compounds-gas forming agents (such as carbonate or bicarbonate salts with/without tartaric acid and/or citric acid).8 Polymers are of great importance for drug delivery systems. In this study, effervescent and non-effervescent floating tablets were studied using different polymers like HPMC, NaCMC, and SA, both gel-forming and swellable hydrophilic polymers. When these polymers come into contact with gastric fluid, they become hydrated and form a colloidal gel barrier on the surface. This diffusional barrier controls the transition of gastric fluid into the tablet and drug molecules release. While the polymer on the outer surface of the tablet dissolves over time, the inner gel layer is protected by the hydration of the adjacent hydrocolloid layer. Also, the air trapped by the swollen polymer lowers the density of the tablet and creates adequate lift.15,16 HPMC and NaCMC are the semi-synthetic cellulose ether most commonly used in drug formulations for controlled drug release. They are also used as an emulsifying agent, thickener, suspending agent, stabilizer, binder, and film-former in pharmaceuticals and in cosmetics.16-18 Alginate is a naturally occurring anionic polysaccharide isolated from brown marine algae. Alginate is a biocompatible and nontoxic that used as a stabilizer and drug release modifier/extender in pharmaceutical formulations.18,19
In this study, the lyophilization technique was used in the preparation of effervescent and non-effervescent floating tablets. Freeze-drying or lyophilization is a drying technique in which the ice in the frozen product is removed by sublimation. The lyophilization technique has been used in the pharmaceutical industry for years but has gained popularity in preparing orally fast disintegrating/dissolving tablets (orodispersible tablets) and films in recent years. Tablets prepared with this technique are highly porous and of low density, allowing body fluids to penetrate rapidly into the tablet and dissolve quickly in the body. Also for freeze-dried films, it provides better folding durability and shorter disintegration time.4,20 In the last few years, this technique has been used to prepare floating tablets, too. There are very few lyophilized floating tablet studies in the literature. The floating tablets prepared by this technique have a highly porous and low-density structure, providing the ability to float without delay, and freeze-drying has an advantageous effect on prolonging gastroretension. Moreover, the freeze-dried tablet also shows good hardness and low friability due to the presence of polymers like HPMC.5,20,21
Antibiotics are often used for stomach or intestinal infections (such as peptic ulcers, ulcerative colitis, small intestine infections, and tuberculosis). Most antibiotics have problems such as being unstable in the stomach pH, remaining in the stomach for a very short time and low antibiotic concentration in the deep stomach mucus layer where bacteria live. In addition, due to their low solubility in the lipid membrane, antibiotics either pass little or not through the cell membrane. In recent years, many controlled and extended-release pharmaceutical dosage forms have been studied to overcome these problems of antibiotics and increase their effectiveness. Gastroretentive drug delivery systems are one of the best examples of these systems.22,23 Ampicillin, a broad-spectrum antibiotic, is widely used locally or systemically for stomach or intestinal infections. Therefore, Amp.Na which has a high water solubility, was preferred as a model drug in this study. Amp.Na has a short biological half-life of 0.74-1.5 hours and is ideal for this system. This study aims to prepare and characterize floating lyophilized tablets, one of the new drug delivery systems, to understand the effect of different biopolymers on quality control parameters more clearly. Within the scope of characterization studies, determination of content uniformity, weight deviation, diameter-thickness, hardness and friability, dissolution rate and kinetics, swelling and floating degree, morphological control, and Brunauer-Emmett-Teller (BET), X-ray diffraction (XRD), Fourier transform infrared (FTIR) analyzes were performed.
Materials. Amp.Na and HPMC K100 were a gift from İ.E. Ulagay İlaç Sanayii A.Ş. (Turkey) and Santa Farma İlaç Sanayii A.Ş. (Turkey). NaCMC (Mw ~700000), SA (low viscosity) and Avicel RC 581 were purchased from Sigma (Germany), Alfa Aesar® (Germany) and FMC Biopolymers (Belgium), respectively. Calcium chloride (CaCl2) and calcium carbonate (CaCO3) were purchased from J. T. Baker (Holland). For pH 1.2 hydrochloric acid (HCl) buffer (USP30-NF25), HCl and potassium hydroxide were purchased from Isolab (Germany) and Riedel-de Haën (Germany). For simulated gastric fluid, sodium chloride (NaCl) and pepsin (from porcine gastric mucosa, ≥500 IU/mg) were purchased from J. T. Baker (Holland) and Sigma (China). All chemicals used were of analytical or pharmaceutical grade. Ultrapure water was used in all studies (Merck Millipore Direct-Q™ 3, Germany).
Preparation of Floating Lyophilized Tablet Formulations. The lyophilization technique was used in the preparation of floating tablets. In the preliminary studies, the volume of the blister to be used was determined first, and the amount of polymer and active substance required for a tablet in accordance with this volume was determined by taking into account the swelling rates of the polymers. First, Amp.Na (0.25 g) and a small amount of CaCl2 as crosslinker were dissolved in ultrapure water homogeneously on a multi-point magnetic stirrer (2mag, MIX 15 eco, Germany). Then the water-soluble/swellable polymers (HPMC K100 single and/or with NaCMC or SA) were dissolved/swelled in this solution. Avicel RC 581 was added to this prepared dispersion as a matrix former-thickener and made homogeneous by stirring. For effervescent floating tablets, CaCO3 was added as a gas-forming agent at the last step. This dispersion was then filled into suitable tablet blisters with the aid of a spatula and lyophilized for 24 hours (CHRIST Alpha 2-4 LSC plus, Germany) after freezing overnight at -20 °C (minimum n=30). Amp.Na-free (blank) floating tablets were also prepared without the Amp.Na addition only, as described above.5,21
In vitro Characterization of Floating Lyophilized Tablet Formulations. Determination of Floating Degrees of Lyo- philized Tablet Formulations: The floating degree determination of randomly selected lyophilized tablets with Amp.Na was performed using 50 mL pH 1.2 HCl buffer in 100 mL beakers at a temperature of 37±0.5 °C and 50 rpm in a horizontal shaker water bath (Memmert, WNB 14, Germany). Whether the tablets swelling over time were floating in the medium at certain time intervals (0, 0.5, 1, 2, 4, 8, 24 hours) were evaluated for 24 hours, and the results were determined as floated or not floated.24 Other experiments were continued with lyophilized tablets showing the ability to float for 24 hours.
Determination of Content Uniformity of Floating Lyo- philized Tablet Formulations: Randomly selected floating lyophilized tablets with Amp.Na were crushed in a mortar and stirred in pH 1.2 HCl buffer (50 mL) for 21 hours on a multipoint magnetic stirrer at 1000 rpm. After the tablets were completely disintegrated and the active substance was released, pre-filtering was performed using filter paper in order to remove the tablet residues. Then, supernatants were filtered using a 0.45 µm membrane filter (Agilent, USA) again and Amp.Na was quantified by using the previously validated UV-VIS spectrophotometric method (205 nm, R2=0.9998, y= 0.0462x+0.0162) (Beckman Coulter DU 730, USA). Dilutions, if required, were made with pH 1.2 HCl buffer (minimum n=3).25 The assay of the tablets should be within the specified 90-120% limits of the USP30-NF25 monograph for Amp.Na tablets.26 It has been paid attention that the difference in the content uniformity of the tablets is not more than (-10)-(+20)%. Results are given as mean and standard deviation (SD).
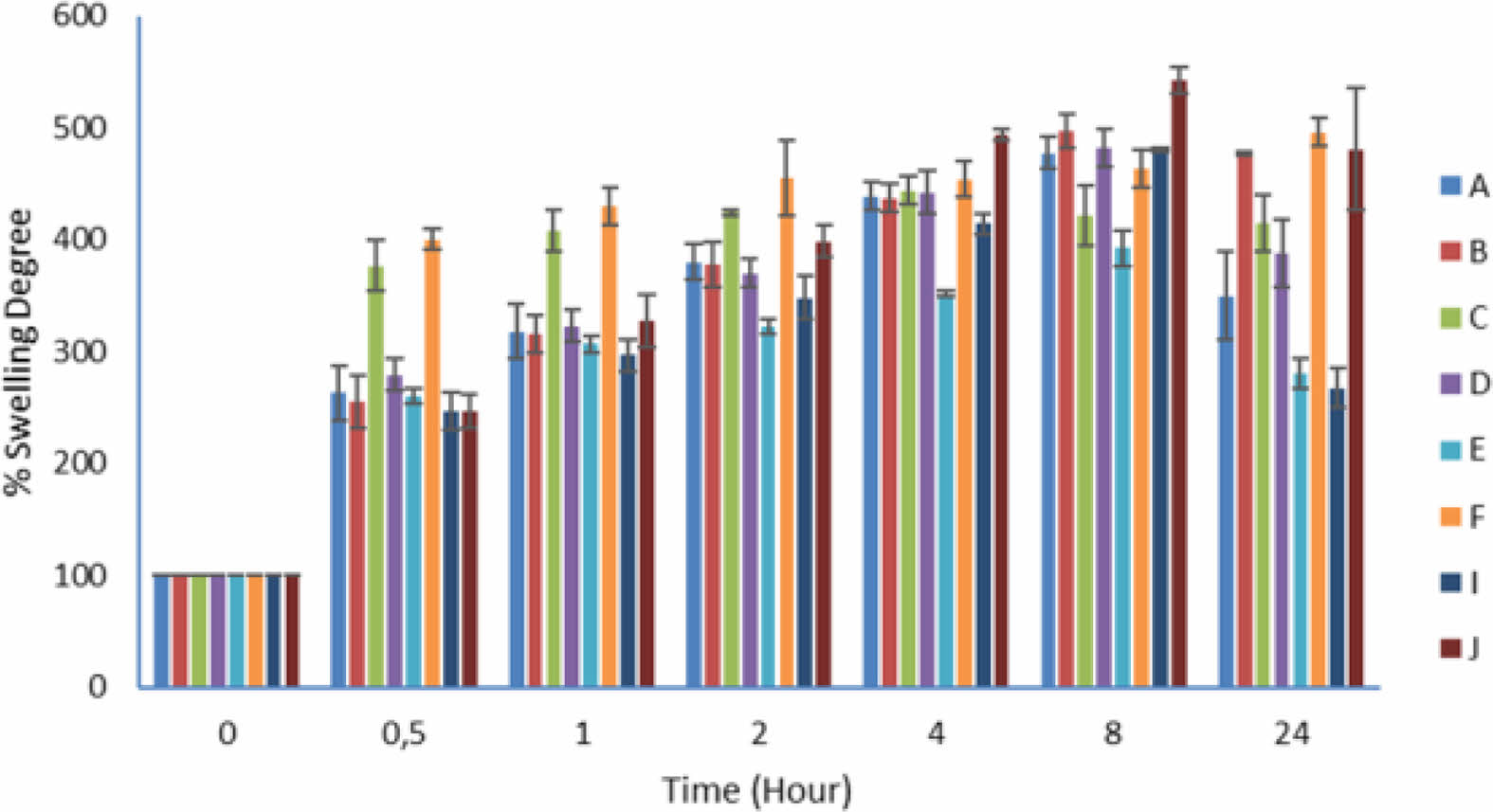
Determination of Swelling Degrees of Floating Lyophilized Tablet Formulations: In order to determine the swelling degree, randomly selected floating lyophilized tablets with Amp.Na were weighed and carried out in 50 mL pH 1.2 HCl buffer using a horizontal shaker water bath at 37 °C and 50 rpm. The tablets that swelled over time were weighed by filtering through the filter at certain time intervals (0, 0.5, 1, 2, 4, 8, 24 hours). The weight before the test was compared with the weight resulting from swelling over time, and the degree of swelling was expressed in % (minimum n=3). The percentages of swelling degree were calculated by using the formula below:27
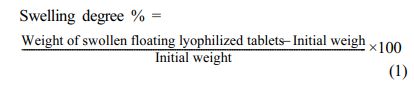
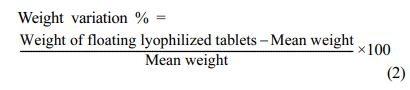
Determination of Diameter, Thickness and Weight Vari- ation of Floating Lyophilized Tablet Formulations: The diameters and thicknesses of randomly selected floating lyophilized tablets with Amp.Na were measured using a manual vernier caliper one by one (n=20). Average and standard deviation values were calculated.28 For weight variation, randomly selected floating lyophilized tablets with Amp.Na each individually weighed on the calibrated analytical balance (n=20). The average weight of the tablets and the deviation values of each tablet from the mean were found. The deviation in the weight of more than two tablets should not be at least 5% or more than 10% in any tablet, as reported in the European Pharmacopoeia (EP 8.0).29 The percent weight variation was calculated by using the formula below:
Determination of Hardness of Floating Lyophilized Tablet Formulations: The hardness of randomly selected floating lyophilized tablets with Amp.Na were measured using a hardness tester (Pharma Test PTB, Germany) (n=10). Results are given as mean and SD.29
Determination of Friability of Floating Lyophilized Tablet Formulations: For the friability testing of floating lyophilized tablets with Amp.Na, 20 randomly selected whole tablets with a unit mass of less than 650 mg were taken. The tablets were carefully dedusted prior to testing and accurately weighed, then placed in the drum. The weighed tablets were rotated for 4 minutes at 25 rpm per minute in the friabilitor (Pharma Test PTF E/ER, Germany). Then, any loose dust from the tablets was dedusted and accurately weighed again. The friability value should not exceed 1%. If the weight loss is more than 1% or the results are doubtful, the test is repeated 3 times and averaged as reported in the EP 8.0.29 The percent friability was calculated by using the formula below:
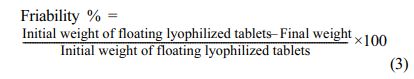
In vitro Release Study and Determination of Release Kinetics of Floating Lyophilized Tablet Formulations: In order to determine the release, the dissolution study of floating lyophilized tablets containing 250 mg Amp.Na was carried out in a 900 mL of pH 1.2 simulated gastric fluid with pepsin (SGF, USP30-NF25) using USP Type II Apparatus (Paddle Type, Pharma Test PTWS IIIE/CE, Germany) at 37±0.5 °C at 100 rpm for 24 hours. An appropriate amount (10 mL) of samples were withdrawn at certain time intervals (0.5, 1, 2, 4, 8, 24 hours) and subsequently, the same amount of fresh pH 1.2 SGF was added to the samples to maintain the sink conditions. Samples were filtered through 0.45 µm membrane filters and released Amp.Na amounts were determined by using the previously validated UV-VIS spectrophotometric method. Dilutions were made with pH 1.2 SGF (n=3). The formula of SGF used was26:

Release results in pH 1.2 SGF were applied to the computer program in order to determine the kinetic model of the release from floating lyophilized tablets with Amp.Na. Whether formulations are compatible with Zero-Order, First-Order, Higuchi, or Korsmeyer-Peppas kinetic models were determined by mathematical operations and formulas.30 The release data obtained were fitted into the following eqs.;
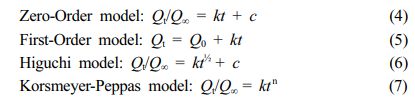
Where, Qt is the released amount of Amp.Na at time t, Q0 is the initial amount of Amp.Na in the tablet, Q∞ is the released amount of Amp.Na at infinite time, k is the release constant of each model, n is the exponent and c is an intercept.31,32 In addition, at the end of this study, the floating ability after 24 hours was also evaluated in pH 1.2 SGF.
Determination of Surface Area and Pore Diameter Analysis of Floating Lyophilized Tablet Formulations: The surface areas and average pore diameters for floating lyophilized tablets with Amp.Na were obtained via the BET method by using nitrogen adsorption isotherms using a Micromeritics 3Flex (USA) analyzer. The system temperature was maintained at 77 K.33 This study was performed at East Anatolian High Technology Research and Application Center (DAYTAM) of Atatürk University.
Fourier Transform Infrared (FTIR) Spectroscopy Study of Floating Lyophilized Tablet Formulations: Powder samples of Amp.Na and floating lyophilized tablets were prepared to determine if there were any unwanted interactions between the formulations and the active ingredient. Infrared spectra were taken by FTIR (ATR) spectrometer (Bruker VERTEX 70v, USA) directly over the powder sample in the range of 4000-400 cm-1 wavenumber.33,34 This study was performed at East Anatolian High Technology Research and Application Center (DAYTAM) of Atatürk University.
X-ray Diffraction (XRD) Study of Floating Lyophilized Tablet Formulations: The crystallinities of Amp.Na and floating lyophilized tablets were determined by the XRD method. XRD studies were carried out by using an X-ray diffractometer (PANalytical Empyrean XRD) fitted with a copper target, a voltage of 45 kV, and a current of 40 mA.35 This study was performed at East Anatolian High Technology Research and Application Center (DAYTAM) of Atatürk University.
Statistical Analysis: Release data were compared by one-way analysis of variance (ANOVA) with LSD using SPSS version 13.0 (SPSS Inc., Chicago, USA). The p values were calculated, and p<0.05 was considered as significant.
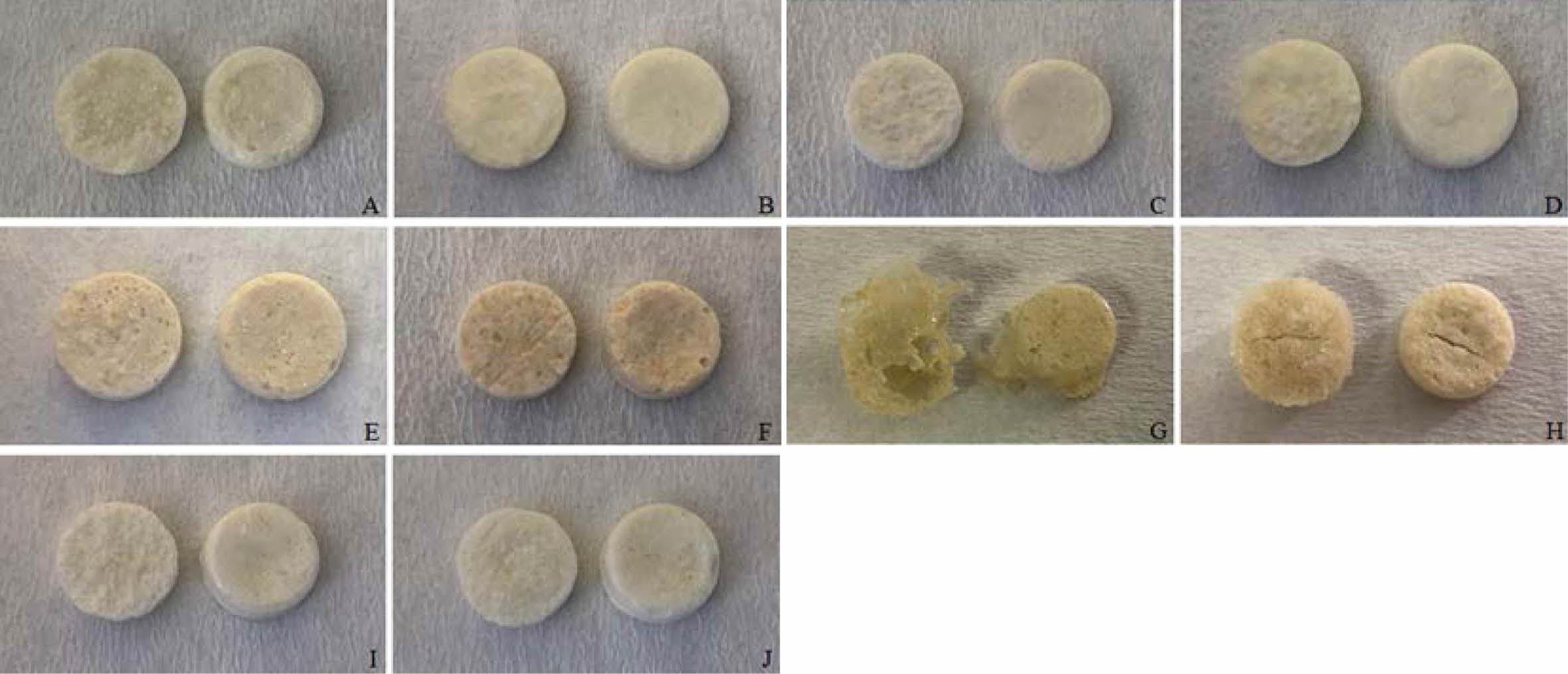
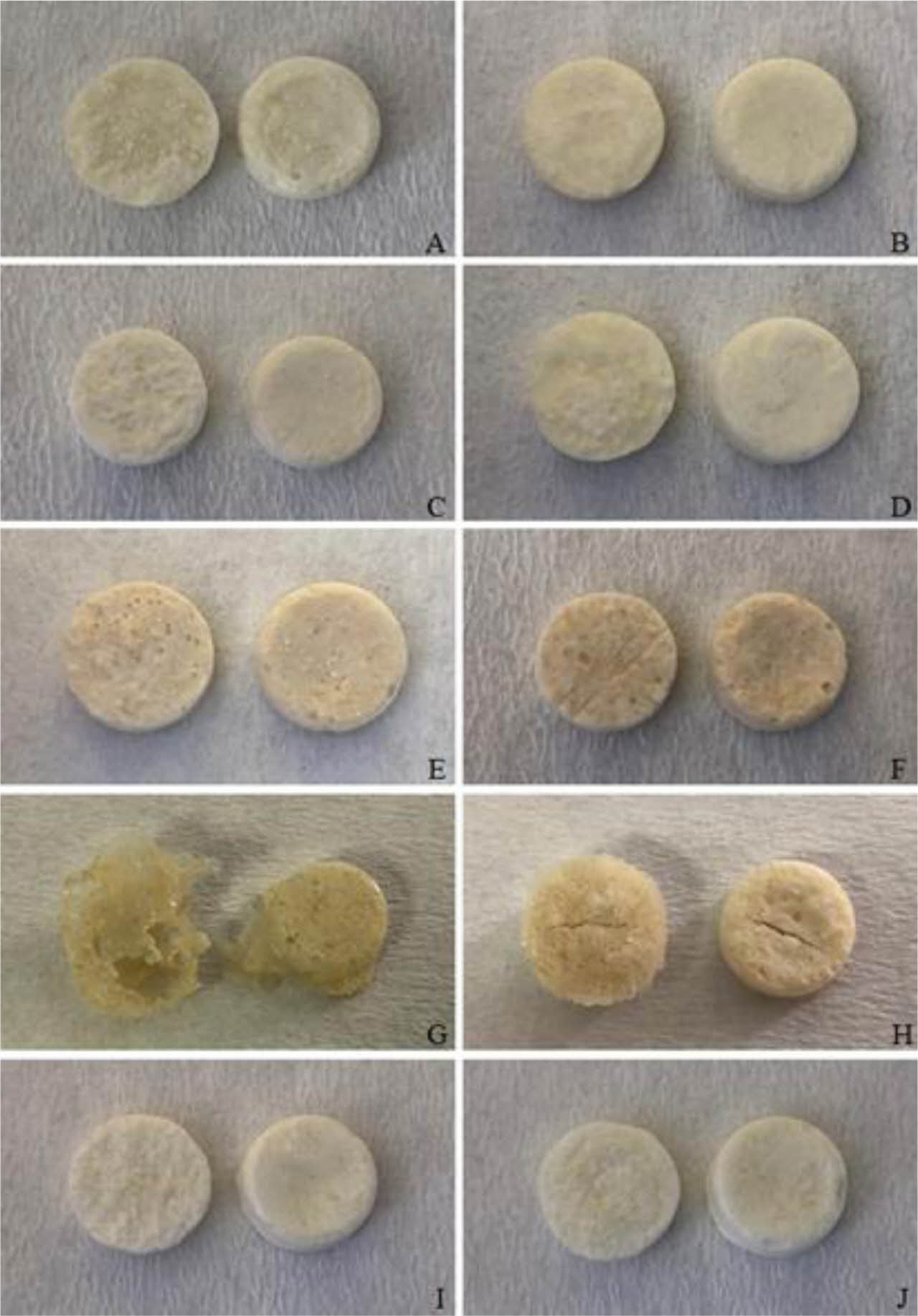
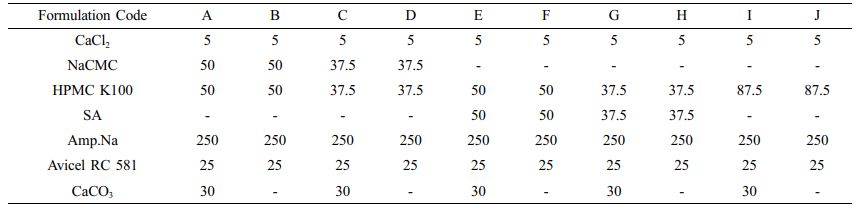
Preparation of Floating Lyophilized Tablet Formulations. Floating lyophilized tablets were successfully prepared by lyophilization technique using three different polymers (HPMC K100 single and/or with NaCMC or SA). The lyophilization technique has become highly preferred in recent years in the development of non-hygroscopic and highly porous tablets with sufficient strength, improved disintegration time, and acceptable quality characteristics of the prepared tablets.21,36 The comparison of prepared tablets formulations is given in Table 1. Amounts are given in mg. As a result of the pre-formulation studies, different combinations were created by changing the formulation components (CaCl2: cross-linker, NaCMC, HPMC K100, SA: water-soluble/swellable polymers, Amp.Na: model drug, Avicel RC 581: matrix former, CaCO3: gas-forming agent). Formulations A, C, E, G and I were prepared as effervescent floating tablets with CaCO3, and formulations B, D, F, H and J were prepared as non-effervescent floating tablets without CaCO3. When the formulations were examined in terms of shape, only the formulations G and H could not reach the desired result. After the lyophilization, foaming occurred in formulation G, and mid-breaking occurred in formulation H, so they could not show optimum tablet properties. For this reason, these formulations (G and H) were canceled, and all studies have been done with the other formulations (A, B, C, D, E, F, I and J). When evaluated in terms of formulation components, it was thought that CaCO3 in formulation G also increases foaming with insufficient polymer amount. In addition, HPMC K100 and SA in combination in the formulations G and H are less in terms of polymer amount compared to the formulations E and F. These small amounts of polymers did not provide sufficient matrix structure and strength to the tablets. In addition, when the formulations are evaluated in terms of color, formulations E, F, G and H are yellowish in color due to the use of SA. In contrast, the others are white in color. The images of all the tablets obtained are shown in Figure 1.
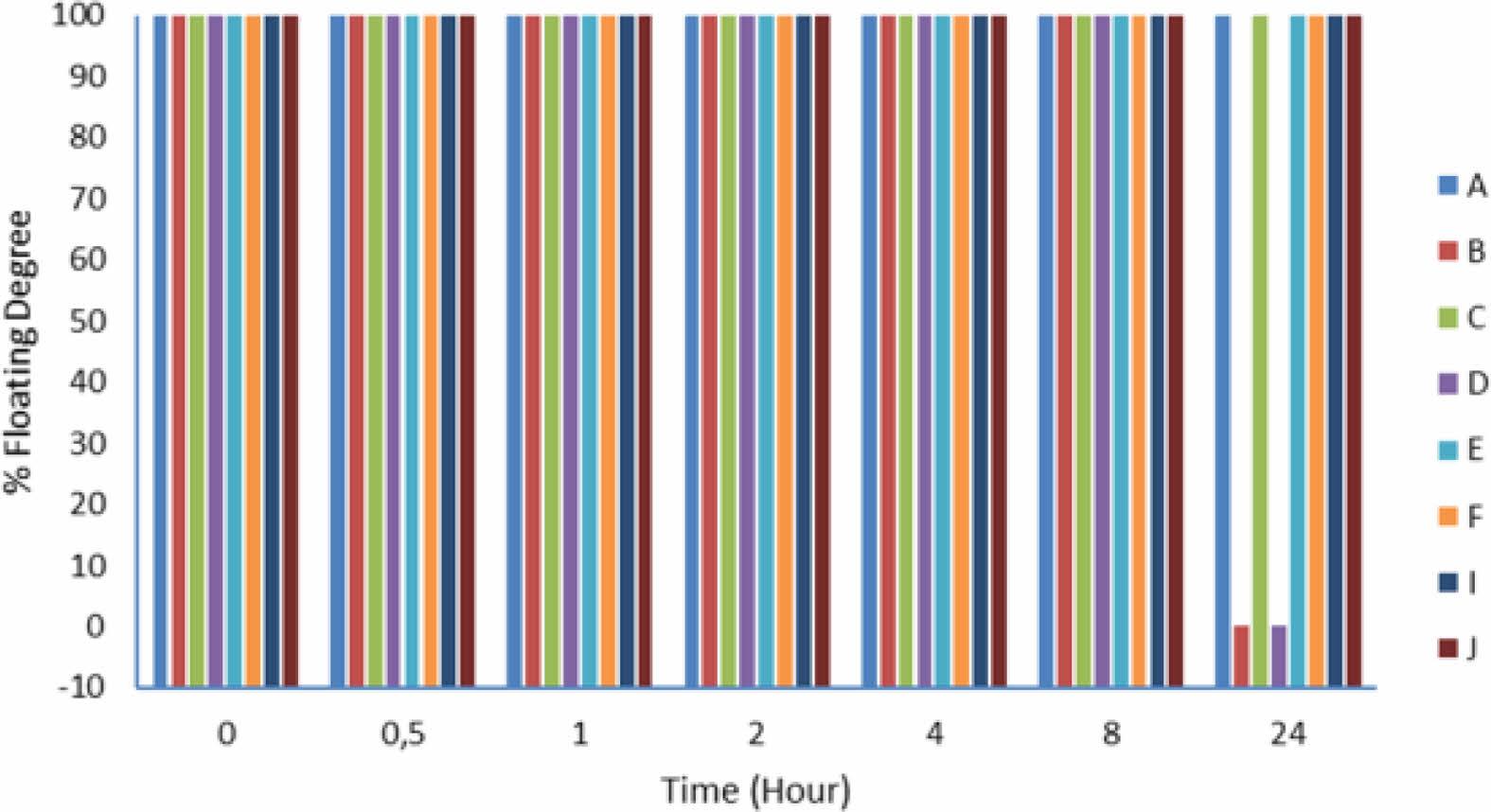
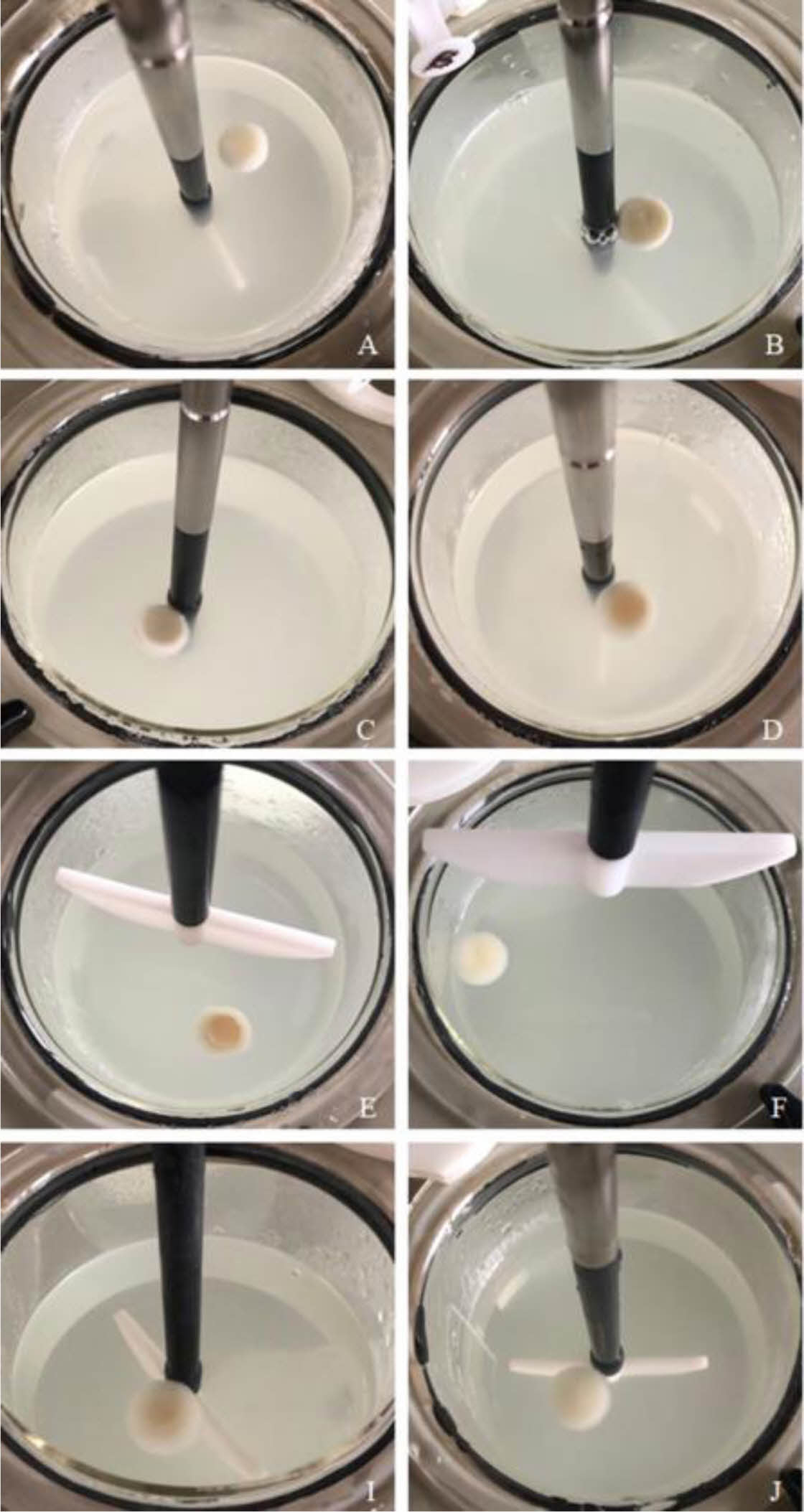
In vitro Characterization of Floating Lyophilized Tablet Formulations. Determination of Floating Degrees of Lyo- philized Tablet Formulations: The floating study was successfully performed in pH 1.2 HCl buffer at 37 °C for 24 hours. The obtained results are shown in Figure 2 in graphic form, and the tablet images after 24-hours are shown in Figure 3. As a result of this study, only formulations B and D were unsuccessful, while other formulations did. As seen in the figure, formulations B and D collapsed with their structural integrity intact. When evaluated in terms of formulation content, NaCMC and HPMC K100 in combination were used as polymers, and CaCO3 was not used in both formulations B and D because they were non-effervescent floating tablets.
When formulations B and D were compared with formulations A and C, the absence of CaCO3 in their structure prevented them from floating. Especially for these two formulations, the presence of CaCO3 is critical because the polymers in the structure were not enough for floating. CaCO3, which creates gas (CO2) in the acidic buffer medium, leads to the formation of pores in the matrix and enables the density to decrease below 1 g/mL and float.15,37 The reaction of CaCO3 with HCl takes place as shown below38:
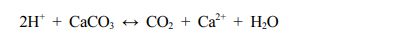
It is known that the amount of gas-forming agent (calcium carbonate or sodium bicarbonate) of 5-50% of the total amount in floating dosage forms will be beneficial for floating. When all our formulations are examined in this respect, it is seen that the calcium carbonate amounts are more than 5%.18 In addition, the other ingredients and their amounts used in the formulation seem ideal in terms of maintaining the tablet’s integrity. Except for those, formulation E disintegrated but did not collapse. Formulation E was prepared with HPMC K100 and SA compared to all other formulations. We see that CaCO3 is used together with HPMC K100 and SA in this formulation. SA tended to disintegrate faster in the presence of CaCO3. Here, we can say that CaCO3 accelerates SA disintegration due to the weakening of the polymer chains while forming gas in the buffer medium. In the presence of CaCO3, the pores formed with the outflow of CO2 increase the surface area and also the contact of the polymer with water. Thus, the polymer in contact with water begins to decompose over time. In other words, the main disintegration mechanism is not due to the presence of CaCO3, but to the porous structure formed due to the presence of CaCO3. There are very few studies on the ability of alginates to act as disintegrants in tablets. This feature comes from the highly swellable characteristics of alginate.39-41 If we also evaluate the formulations G and H to formulations E and F, the low amount of SA (37.5 mg) and HPMC K100 (37.5 mg) caused a problem during lyophilization (the amounts of SA and HPMC K100 in formulations E and F are 50 mg each). Here it is understood that the presence of the low amount of SA and the presence of CaCO3 together with the SA cause problems when preparing the formulation or when trying to float. In a study, Jin et al. made wound dressings using hydrophilic polymers on wound healing and used SA, NaCMC, HPMC, PVP, PVA, and poloxamer as polymers. In their experiment to compare the degree of swelling of the polymers, they found that the polymer that showed the most swelling was SA, and the order is: SA > NaCMC = poloxamer = HPMC > PVA = PVP.42 From this, we clearly understand that as a result of the rapid swelling of SA, the polymer chains begin to loosen and break apart but do not lose their buoyancy.
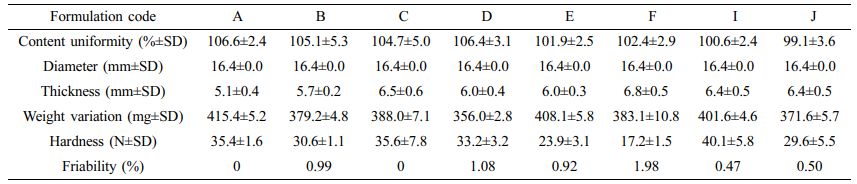
Determination of Content Uniformity of Floating Lyophilized Tablet Formulations: The content uniformity study was successfully performed in pH 1.2 HCl buffer at 37 °C for 21 hours. The content uniformity results are given in Table 2. All the prepared formulations did not exceed the 10% limit, and the values were found in the range of 99.1±3.6-106.6±2.4%.
Determination of Swelling Degrees of Floating Lyophilized Tablet Formulations: The swelling study was successfully performed in pH 1.2 HCl buffer at 37 °C for 24 hours. The results of the swelling degrees are given in Table 3 below (mean±SD), and the graph is shown in Figure 4. Cellulose-based polymers and sodium alginate are polymers with a long chain structure. Due to the strong hydrophilicity of these polymers, hydrogen bonds formed between water molecules and polar groups (including OH and COO-) along with the polymer. Hydrogen bonds are formed both between the same chains of these polymers (intra-molecular) and between neighboring chains (inter-molecular). These strong hydrogen bonds make it difficult for polymers to dissolve in aqueous mediums. However, breaking these strong bonds causes the polymers to swell.43,44 When the tablets come into contact with the buffer medium, the hydrogen bonds begin to break, and then swelling occurs. Thus a loose and thick gel layer is formed on the tablet surface, which erodes for a certain period of time and facilitates the release of the active substance.30,45 The data obtained in the floating study images and the swelling study confirm this information. All of our tablets first swelled and then gradually began to disintegrate from the outside. In addition, the degree of swelling is a sign of the hydrophilicity of the polymers. SA, NaCMC and HPMC with more hydrophilic functional groups, such as carboxyl and hydroxyl groups, have more swellable characteristics.42
When formulations A and B were examined, NaCMC and HPMC K100 were used in combination, and only formulation A contained CaCO3. Formulations A and B showed similar swelling properties in the first 8 hours of swelling time, but formulation A showed more erosion at 24 hours, while formulation B did not show much erosion. We can say that this is due to CaCO3 in formulation A.
Again, when we examined the formulations C and D, CaCO3 in the formulation C accelerated the swelling with this mechanism of action. The formulations C and D erosion has increased in the hours after the 4th and the 8th hours, respectively. Comparing formulations C and D with formulations A and B, the amount of polymer is lower. This situation indicates the presence of a looser polymer chain and also causes faster swelling in the first hours. When the formulations E and F were examined, HPMC K100 and SA were used in combination. Here, CaCO3 accelerated the swelling and erosion of formulation E at the same time (8th hours) like formulations A and C, and so, the swelling degree of formulation E was not increased like the other formulations. Since the presence of SA in formulation E did not create sufficient strength like other polymer combinations, the lowest swelling rate was observed in formulation E after 8 hours. However, the most effective situation here is the polymer erosion with swelling. Formulations I and J were prepared using HPMC K100 only. Formulation I containing CaCO3 behaved like formulations A, B, C, and D in the first 4 hours but started to erode rapidly after 8 hours and was the formulation showing the most erosion among all formulations at 24 hours. We can say that this is caused by the highly hydrophilic nature of HPMC K100 and the presence of CaCO3. Contrary to this situation, formulation J shows the highest swelling rate from the 4th hours to 8th hours compared to all other formulations. Since CaCO3 is not used in this formulation and it consists only of HPMC K100 (the quantity of HPMC K100 is the highest), it is seen that swelling predominates to erosion.37,46
Determination of Diameter, Thickness and Weight Variation of Floating Lyophilized Tablet Formulations: The diameter, thickness and, weight variation study were successfully performed. The results are given in Table 2 as mean±SD. The diameters of all formulations are 16.4±0.0 mm. The thicknesses of all formulations are in the range of 5.1±0.4 mm and 6.8±0.5 mm. The weight variations of all formulations are in the range of 356.0±2.8-415.4±5.2 mg, that the weight deviations are less than 5%. That is, all the tablets meet pharmacopoeial (EP 8.0) requirements.
Determination of Hardness of Floating Lyophilized Tablet Formulations: The hardness study was successfully performed. The results are given in Table 2 as mean±SD. The hardnesses of all formulations are in the range of 17.2±1.5-40.1±5.8 N.
Determination of Friability of Floating Lyophilized Tablet Formulations: The friability study was successfully performed. The results are given in Table 2 as mean±SD. Except for formulation F only, other formulations passed the friability test; that is, they have withstood mechanical stress. When the hardness and friability values of all formulations are compared, it is seen that the formulation with the lowest hardness value is the F formulation. Compared to the tested formulation E, the difference is only CaCO3 present in the E formulation. Here, the amount of CaCO3 in the tablet matrix gave the formulation sufficient hardness to pass the test. We can say that CaCO3 is a water-insoluble substance, and it adds strength to the structure by filling in between the polymer chains in the environment. In addition, if we evaluate in terms of thickness, we see that the thickest tablet is the formulation F. We can also say that the abrasion is high because the surface area to be eroded by hitting the surface in the test device increases due to the increase in the thickness as the thickness increases.
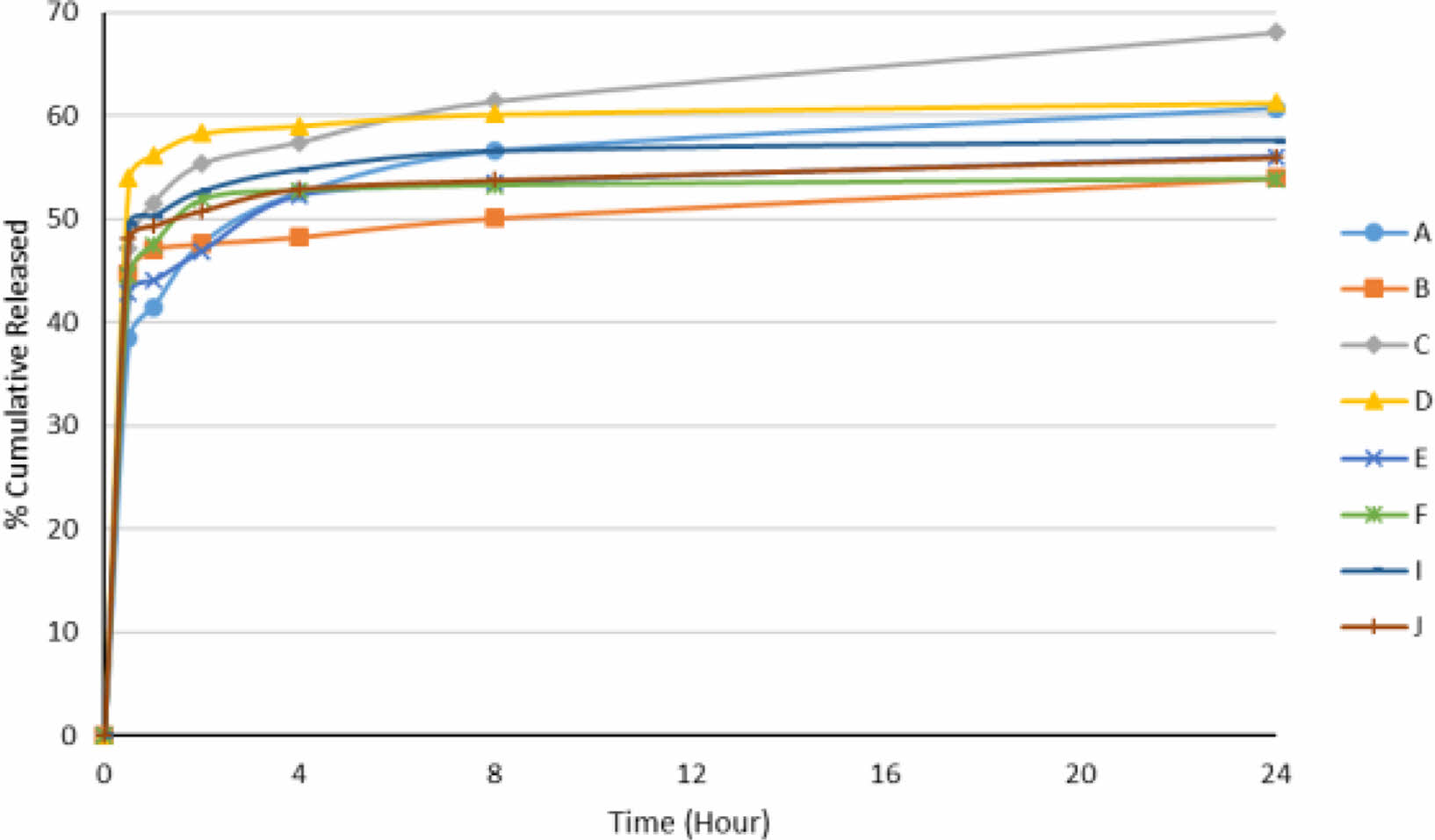
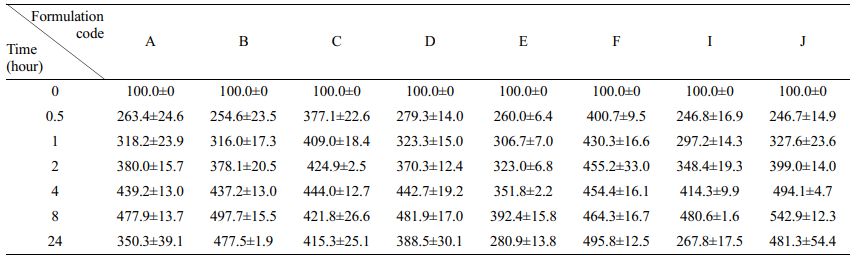
In-vitro Release Study and Determination of Release Kinetics of Floating Lyophilized Tablet Formulations: The release study was successfully performed in pH 1.2 SGF with pepsin at 37 °C and 100 rpm for 24 hours using USP Type II Apparatus. In the release study, pH 1.2 SGF with pepsin was used to evaluate the release and buoyancy properties of the tablets in the presence of enzymes. The cumulative release results are shown in Figure 5 below (mean±SD).
When the release profiles of both effervescent and non-effervescent floating tablets were examined, they all showed similar release profiles. When the release amount of all formulations in the first half-hour was examined, the lowest 38.45% (formulation A) and the highest 53.94% (formulation D) release were observed. In general, we can say that the burst effect is caused by the presence of pepsin with SGF,47 the hydrophilic structure of Amp.Na and the polymers is seen in all of them in the first half-hour. It is seen that 50% release values are reached at the latest in formulation B (8th hour) and earlier than the 4th hour in all other formulations. We can say that this slow release is caused by the high amount of polymer in formulation B (100 mg in total)48 and the absence of CaCO3. Here, HPMC K100 swells in the pH 1.2 SGF and forms a highly viscous gel structure on the tablet surface, causing the release to slow down.48 A similar situation was seen in a study by Shinde et al., and increasing the amount of HPMC K100M caused the release to slow.49 When the total release values at 24 hours were examined, the fastest release was seen in formulation C, and the slowest release was seen in formulations B and F. The release of formulation C is significantly different from the release of other formulations (p<0.05). However, @100% release was not seen in any formulation. We think that this situation is caused by any tablet's structural integrity is not disrupted during the release. Thus, since disintegration does not occur because the polymer chains in the tablet center do not loosen very quickly with the diffusion of water, the release slows down and prolongs.50 It is also thought that the presence of pepsin in SGF initially does not affect the rapid tablet surface release of Amp.Na but slows the release of Amp.Na near the tablet center in the following hours due to its absorption into the hydrogel matrix surface. A similar situation was observed in the riboflavin-containing chitosan and chitosan-PEO hydrogels made by Patel et al., and the presence of pepsin/pancreatin slowed riboflavin release in all hydrogels.51
It was also evaluated whether the tablets floated after 24 hours of release, and all tablets were found to float at pH 1.2 SGF after release. Digital images are shown in Figure 6. In the floating study with pH 1.2 HCl buffer, disintegration was observed in formulation E. In contrast, in the release study with pepsin containing SGF, no degradation was observed in any formulation during 24 hours. As a possible explanation for this situation, we can say that the enzyme has a more negligible effect on the relaxation of the polymers' chains with less HCl in SGF.52 Formulations A and C are effervescent floating tablets, and it is seen that formulation C has less polymer content (total 75 mg) than formulation A. This situation allows to have a looser polymer chain network and accelerates the release. The release is slow as there is more polymer in formulations A and B, and therefore, a tighter network of polymer chains. However, when looking at all formulations in general, effervescent floating tablets released more than non-effervescent floating tablets, so the presence of CaCO3 affected the release. The release of formulation A (60.76%) was significantly different from the releases of formulations B, C and F (p<0.05). Again, no significant difference was found with formulation D as it showed a similar release with formulation D (61.27%) (p>0.05). When the other four formulations (E, F, I and J) are examined, it is seen that there is not much difference between them (p>0.05). The polymers in their structures, the amount/number of polymers, and the presence/absence of CaCO3 did not make a big difference. Non-effervescent formulations B and F showed the same release profile in the first 1 hour and 24 hours. In these two formulations, NaCMC (50 mg) and SA (50 mg), which are out of HPMC K100 (50 mg), did not make a significant difference on the release even in the presence of CaCO3 (formulations A and E) (p>0.05). As a result, there are effects of the amount of polymer, the type of polymer, the swelling of the polymer to form a dense gel barrier, the presence of CaCO3, the diffusion of the drug, and the polymer erosion on drug release.49
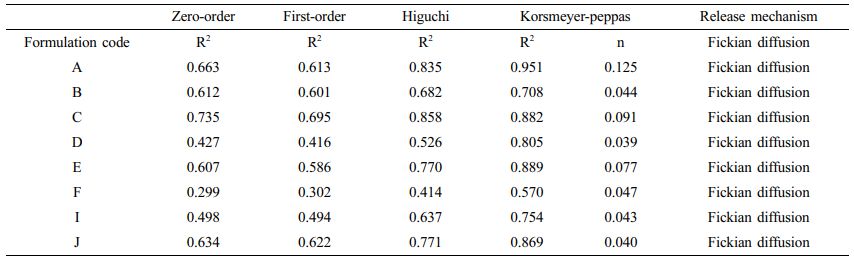
In vitro drug release data in pH 1.2 SGF of all the floating lyophilized tablets were subjected to the goodness of fit test by linear regression analysis according to Zero-Order, First-Order, Higuchi, or Korsmeyer-Peppas kinetic models to ascertain the mechanism of drug release. The results of release kinetics are given in Table 4. In the evaluation made on the value of R2, the highest values in all formulations are seen in the Korsmeyer-Peppas model. The value of “n” is used to characterize the drug release mechanism. According to the limits of the Korsmeyer-Peppas model, which explains the drug release behavior for cylindrical shaped dosage forms such as tablets and formed gel patches, n values <0.45 indicate Fickian diffusional release. If the value of “n” is 0.45-0.89 shows non-Fickian (anomalous) diffusion.50,53 So, this model has shown that all of the formulations’ drug release is Fickian diffusion-controlled.
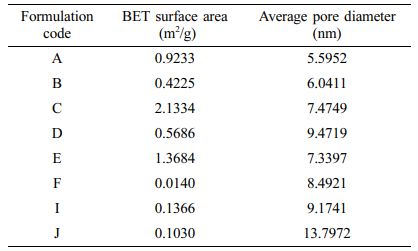
Determination of Surface Area and Pore Diameter Analysis of Floating Lyophilized Tablet Formulations: The pore characteristics of the floating lyophilized tablets are given in Table 5. BET surface areas and average pore diameters of the formulations are given. The surface areas of the formulations are in the range of 0.0013-2.1334 m2/g, and formulation C has the largest surface area. It is recorded in the literature that the surface area and the release influence each other.54 Judging from the release data, it can be said that formulation C has the highest release value, and this is related to its high surface area.
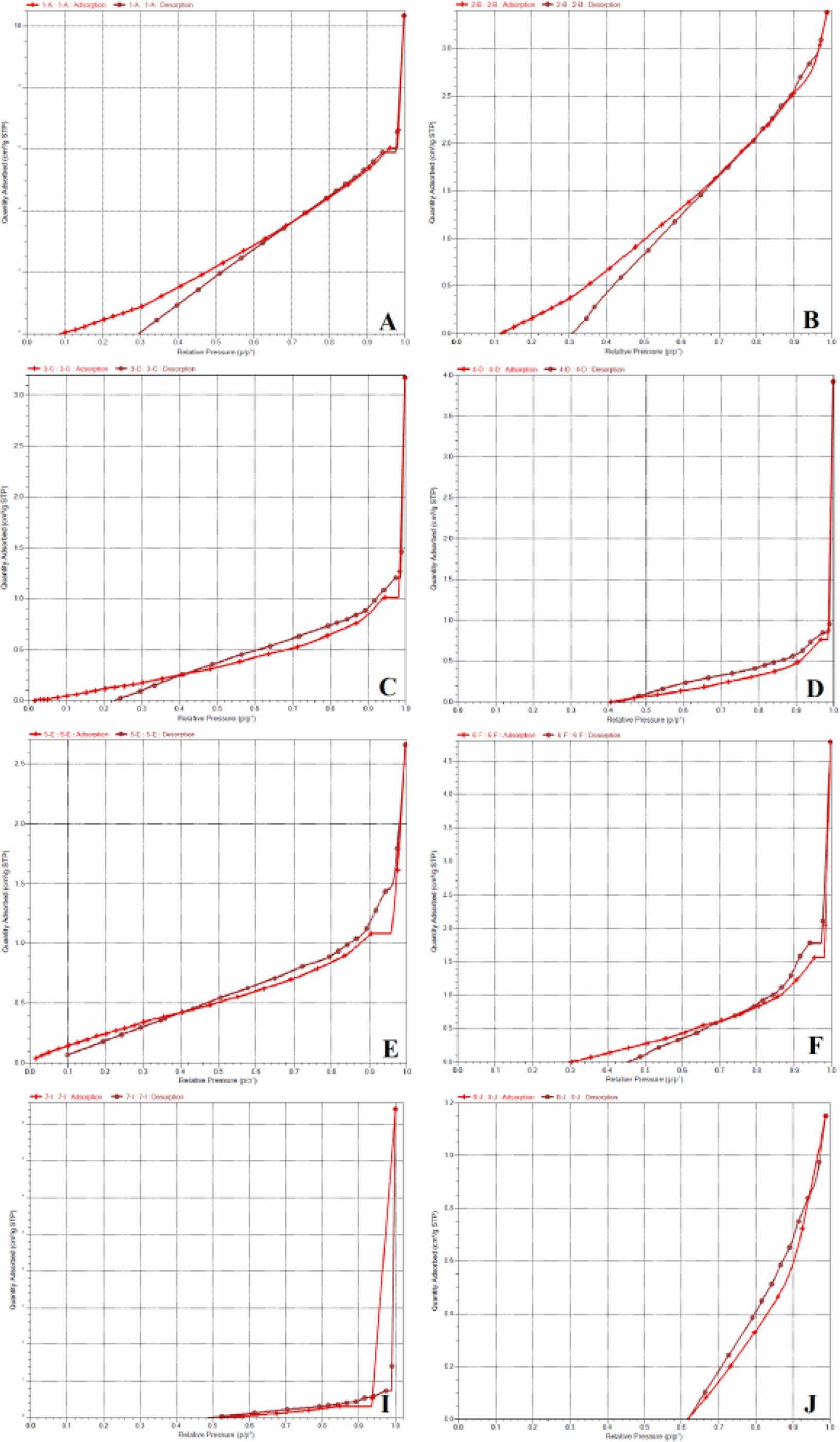
The surface area of all the effervescent floating tablets was greater than the non-effervescent ones. Also, when the pore diameters were compared, the pore diameters of the non-effervescent ones were larger than the effervescent ones. This situation is thought to be caused by CaCO3. From the BET results, it was supported that the presence of CaCO3 caused the CO2 output to increase the surface area and decrease the pore diameters.Figure 7 shows N2 adsorption-desorption isotherms of floating lyophilized tablets. As it is understood from the figures, the isotherms conform to type IV. Type IV shows a hysteresis loop, i.e., the adsorption and desorption isotherms do not coincide over a certain region of external pressures. The type IV isotherm is typical for mesoporous materials. At low pressures, first, an adsorbate monolayer is formed on the pore surface, which is followed by multilayer formation.55 According to the International Union of Pure and Applied Chemistry (IUPAC) nomenclature, pores are roughly classified into the following groups: macropores >50 nm, mesopores in the range of 2.0-50 nm, and micropores <2.0 nm.
The hysteresis loop indicates that the mesoporous structures exist in both floating lyophilized tablets.1 All formulations have pore size distributions around 5.5952-13.7972 nm. However, we can identify all of them as mesoporous.
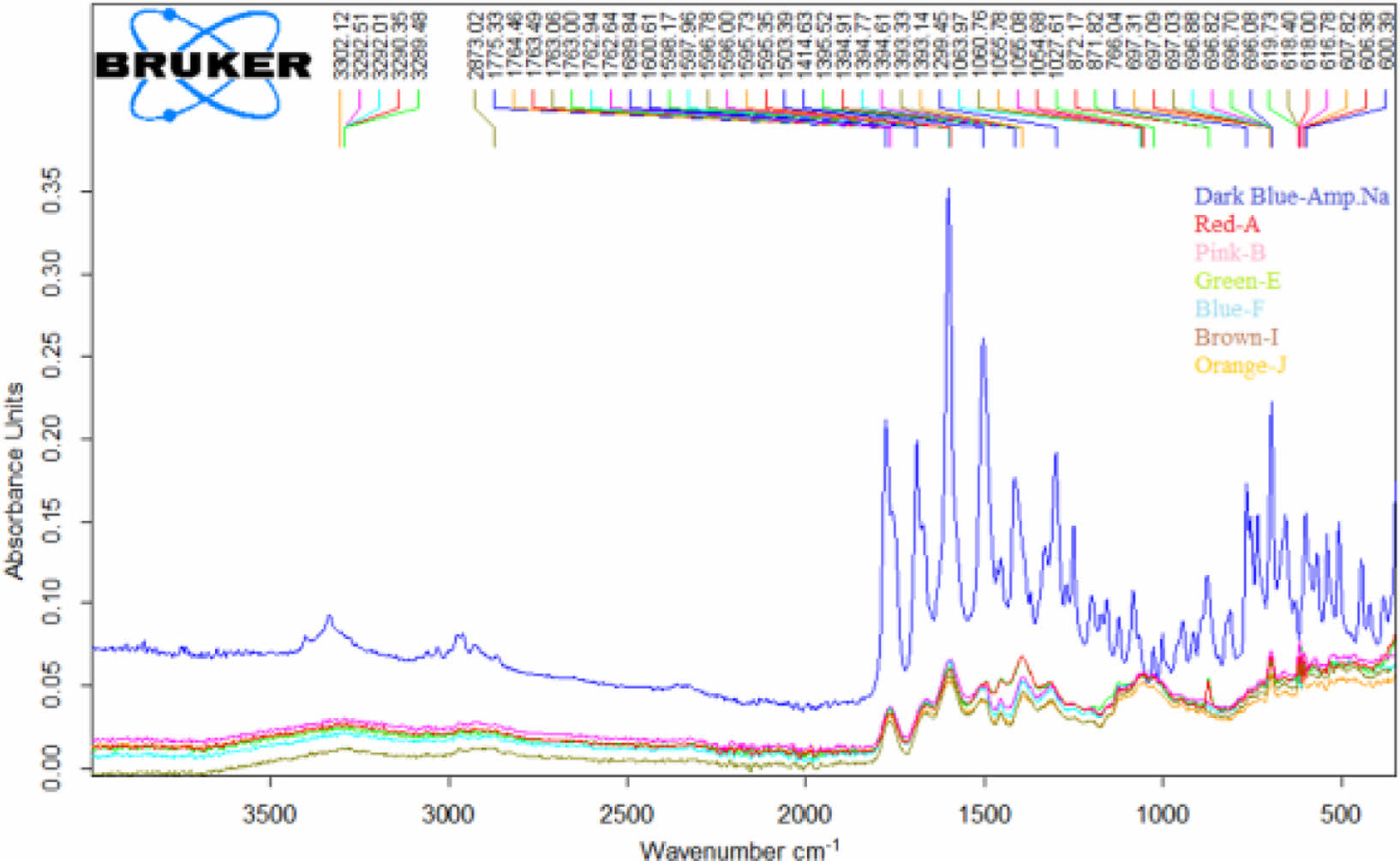
FTIR Study of Floating Lyophilized Tablet Formulations: The FTIR spectra of the Amp.Na and floating lyophilized tablets (except C and D formulations) are shown in Figure 8. Since all the excipients in formulations A and C and formulations B and D are the same, the spectra of the C and D formulations were not taken, and studies were conducted with other formulations. IR spectroscopy examines vibrations caused by infrared rays, stresses of bonds between atoms, and changes in bond angles. When molecules absorb this energy, vibrational energy transitions occur in the infrared region. Given that the molecule absorbs only certain frequencies by infrared rays due to its structure, the vibration frequency is associated with a particular bond type. Therefore, each vibration frequency is unique to that region of that molecule. Consequently, it is a crucial analysis technique and frequently used in detecting changes in molecular structure.56 When the characteristic band voltages of Amp.Na are examined; ~1600 cm-1 (aromatic C=C), ~1500 cm-1 (C-H bending), ~1410 cm-1 (-COOH bending), ~1370 cm-1 (C-H bending), ~1250 cm-1 (C-O strecthing), ~1090 cm-1 (C-O and O-H bending) and ~940 cm-1 (O-H bending) specific peaks were observed.56-58
Some characteristic Amp.Na peaks are also included in the floating lyophilized tablets spectrum. The decrease was observed in sharp peaks of Amp.Na pure peak compared to the tablets peaks due to the polymers. When Amp.Na was treated or formulated with another chemical, specific sharp peaks of Amp.Na were suppressed. This means that Amp.Na is within the tablets, and there was no chemical interaction between the drug and the excipients, so Amp.Na is compatible with the excipients used in the formulations. Similar situations have been observed in other studies as well.57-59 In Vanitha et al. study, the FTIR spectra of pure hydralazine HCl and optimized floating tablet formulation were compared. They stated that specific band peaks of hydralazine HCl were seen in tablet formulation with low intensity, and there was no interaction between chemicals.48
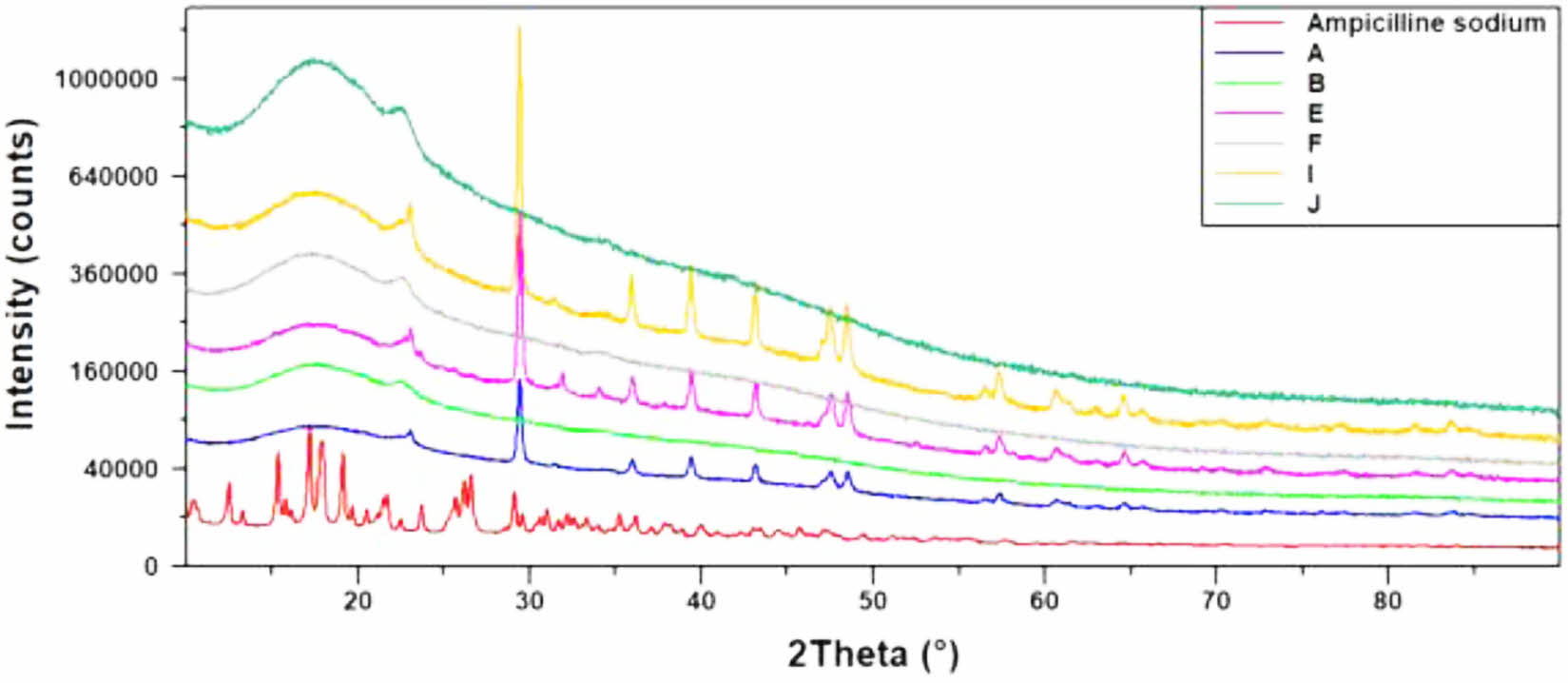
XRD Study of Floating Lyophilized Tablet Formulations: The X-ray diffraction analysis helps to determine the crystal or amorphous nature of the active ingredients in drug delivery systems. X-ray diffractograms of Amp.Na and the floating lyophilized tablets (except formulations C and D) are shown in Figure 9.
Since all the excipients in formulations A and C and formulations B and D are the same, the diffractograms of the formulations C and D were not taken, and studies were conducted with other formulations. Characteristic crystalline peaks of Amp.Na were observed in the diffractogram prominently. The main thermal characteristic sharp peaks of crystalline Amp.Na were recorded between 0-40° 2θ. While Amp.Na showed a dominant crystal phase with sharp and long diffractions in the diffractogram; this situation decreased and suppressed in formulations diffractograms. It can be concluded that Amp.Na is converted to an amorphous form in formulations. Similar situations have been observed in other studies as well.60,61
|
Figure 1 Digital images of floating lyophilized tablets (left: upside, right: downside) |
|
Figure 2 Floating degrees of floating lyophilized tablets. |

|
Figure 3 Digital images of floating lyophilized tablets after the 24- hour floating test |
|
Figure 4 Swelling degrees of floating lyophilized tablets. |
|
Figure 5 Release profiles of floating lyophilized tablets. |
|
Figure 6 Digital images of floating lyophilized tablets after the 24- hour floating test. |
|
Figure 7 N2 adsorption-desorption isotherms of floating lyophilized tablets. |
|
Figure 8 FTIR spectra of Amp.Na and floating lyophilized tablets. |
|
Figure 9 XRD diffractograms of Amp.Na and floating lyophilized tablets. |
|
Table 2 The Physicochemical Characterization of Floating Lyophilized Tablets Formulations |
|
Table 5 BET Surface Areas and Average Pore Diameters of Floating Lyophilized Tablets |
In this study, effervescent and non-effervescent floating lyophilized tablets with Amp.Na were successfully prepared with polymers and excipients by showing the desired quality control and release properties for Amp.Na (the model drug). The lyophilization technique was used successfully for the preparation of the tablets with HPMC K100 single and/or with NaCMC or SA and other excipients. While the lyophilization technique is often used for orally fast disintegrating/dissolving tablets (orodispersible tablets) and films in recent years, its suitability for floating tablets was also supported by this study. The effects of polymers on formulations are discussed in detail, and the quantities, ratios and other properties of polymers affect many parameters. The formulations have been successfully characterized and compared by evaluating many parameters like floating, content uniformity, swelling, diameter, thickness and weight variation, hardness, friability, release and release kinetics. Tablets have mostly passed quality control tests. The tablets have a mesoporous structure as understood from the BET analysis, and this porous structure affected swelling, buoyancy and release. The release characteristic of Amp.Na conforms to the Fickian diffusion mechanism. It has been determined from the FTIR and XRD analyses of Amp.Na that it does not interact with other excipients and is in an amorphous form in tablets. These formulations developed using a model drug can be used for other drugs in the future, and optimum properties can be easily characterized.
- 1. Veronovski, A.; Tkalec, G.; Knez, Z.; Novak, Z. Characterisation of Biodegradable Pectin Aerogels and Their Potential Use as Drug Carriers. Carbohyd. Polym. 2014, 113, 272-278.
-
- 2. Jindal, S.; Jindal, K.; Das Gupta, G.; Garg, R.; Awasthi, R. Gastroretentive Floating Tablets: An Investigation of Excipients Effect on Tablet Properties. Marmara. Pharm. J. 2016, 20, 100-110.
-
- 3. Wang, S.; Wen, H. Y.; Li, P. F.; Cui, M. S.; Sun, W. L.; Wang, H. Y.; Liu, H. F.; Li, S. Y.; Pan, W. S.; Yang, X. G. Formulation and Evaluation of Gastric-Floating Controlled Release Tablets of Ginkgolides. J. Drug. Deliv. Sci. Tec. 2019, 51, 7-17.
-
- 4. Kim, J. Y.; Kim, S. H.; Rhee, Y. S.; Park, C. W.; Park, E. S. Preparation of Hydroxypropylmethyl Cellulose-Based Porous Matrix for Gastroretentive Delivery of Gabapentin Using the Freeze-Drying Method. Cellulose 2013, 20, 3143-3154.
-
- 5. Kim, J. Y.; Seo, J. W.; Rhee, Y. S.; Park, C. W.; Park, E. S. Freeze-Dried Highly Porous Matrix as a New Gastroretentive Dosage Form for Ecabet Sodium: In Vitro and In Vivo Characterizations. J. Pharm. Sci.-Us. 2014, 103, 262-273.
-
- 6. Patil, H.; Tiwari, R. V.; Repka, M. A. Recent Advancements in Mucoadhesive Floating Drug Delivery Systems: A Mini-review. J. Drug. Deliv. Sci. Tec. 2016, 31, 65-71.
-
- 7. Shishu; Gupta, N.; Aggarwal, N. Stomach-specific Drug Delivery of 5-fluorouracil Using Floating Alginate Beads. AAPS Pharmscitech 2007, 8, 48.
-
- 8. Awasthi, R.; Kulkarni, G. T. Decades of Research in Drug Targeting to the Upper Gastrointestinal Tract Using Gastroretention Technologies: Where Do We Stand? Drug. Deliv. 2016, 23, 378-394.
-
- 9. Swain, R. P.; Nagamani, A.; Shankar, P. U. Formulation and Evaluation of Gastro-bilayer Floating Tablets of Ezetimibe as Immediate Release Layer and Atenolol as Sustained Release Layer. Indian. J. Pharm. Educ. 2019, 53, S93-S103.
-
- 10. Tripathi, J.; Thapa, P.; Maharjan, R.; Jeong, S. H. Current State and Future Perspectives on Gastroretentive Drug Delivery Systems. Pharmaceutics 2019, 11, 193.
-
- 11. Arora, S.; Ali, J.; Ahuja, A.; Khar, R. K.; Baboota, S. Floating Drug Delivery Systems: A Review. AAPS Pharmscitech 2005, 6, E372-E390.
-
- 12. Chinthaginjala, H.; Barghav, G. C.; Reddy, C. M.; Pradeepkumar, B.; Ahad, H. A. Formulation and In Vitro Evaluation of Floating Tablets of Dicloxacillin Sodium Using Different Polymers. J. Young Pharm. 2019, 11, 247-253.
-
- 13. Prajapati, V. D.; Jani, G. K.; Khutliwala, T. A.; Zala, B. S. Raft Forming System-An Upcoming Approach of Gastroretentive Drug Delivery System. J. Control. Release. 2013, 168, 151-165.
-
- 14. Singh, B. N.; Kim, K. H. Floating Drug Delivery Systems: An Approach to Oral Controlled Drug Delivery via Gastric Retention. J. Control. Release. 2000, 63, 235-259.
-
- 15. Prajapati, S. T.; Patel, L. D.; Patel, C. N. Polymers for Floating Drug Delivery System. Syst. Rev. Pharm. 2011, 2, 1-7.
-
- 16. Kamel, S.; Ali, N.; Jahangir, K.; Shah, S. M.; El-Gendy, A. A. Pharmaceutical Significance of Cellulose: A Review. Express Polym. Lett. 2008, 2, 758-778.
-
- 17. Li, C. L.; Martini, L. G.; Ford, J. L.; Roberts, M. The Use of Hypromellose in Oral Drug Delivery. J. Pharm. Pharmacol. 2005, 57, 533-546.
-
- 18. Iglesias, N.; Galbis, E.; Romero-Azogil, L.; Benito, E.; Lucas, R.; Garcia-Martin, M. G.; de-Paz, M. V. In-Depth Study into Polymeric Materials in Low-Density Gastroretentive Formulations. Pharmaceutics 2020, 12, 636.
-
- 19. Hariyadi, D. M.; Islam, N. Current Status of Alginate in Drug Delivery. Adv. Pharm. Pharm. Sci. 2020, 2020, 8886095.
-
- 20. Siow, C. R. S.; Wan Sia Heng, P.; Chan, L. W. Application of Freeze-Drying in the Development of Oral Drug Delivery Systems. Expert. Opin. Drug. Del. 2016, 13, 1595-1608.
-
- 21. Ahmed, T. A.; El-Say, K. M.; Hosny, K. M.; Aljaeid, B. M. Development of Optimized Self-Nanoemulsifying Lyophilized Tablets (SNELTs) to Improve Finasteride Clinical Pharmacokinetic Behavior. Drug. Dev. Ind. Pharm. 2018, 44, 652-661.
-
- 22. Zhao, S.; Lv, Y.; Zhang, J. B.; Wang, B.; Lv, G. J.; Ma, X. J. Gastroretentive Drug Delivery Systems for the Treatment of Helicobacter pylori. World J. Gastroenterol. 2014, 20, 9321-9329.
-
- 23. Nishi, K. K.; Antony, M.; Jayakrishnan, A. Synthesis and Evaluation of Ampicillin-conjugated Gum Arabic Microspheres for Sustained Release. J. Pharm. Pharmacol. 2007, 59, 485-493.
-
- 24. Prasanthi, N. L.; Harshita, Y.; Manikiran, S. S.; Ramarao, N. Design and Development of Gastroretentive Tablets of Coccinia grandis leaf extract for treating Helicobacter pylori infection. Asian J. Pharm. 2019, 13, 184-191.
-
- 25. Pawar, H. A.; Gharat, P. R.; Dhavale, R. V.; Joshi, P. R.; Rakshit, P. P. Development and Evaluation of gastroretentive Floating Tablets of an Antihypertensive Drug Using Hydrogenated Cottonseed Oil. ISRN Pharm. 2013, 2013, 137238.
-
- 26. Abernethy, D. R; The United States Pharmacopeia-National Formulary (USP30-NF25); The United States Pharmacopeial Convention Inc.: Rockville, 2007; p 1101.
- 27. Jagdale, S. C.; Patil, S.; Kuchekar, B. S. Application of Design of Experiment for Floating Drug Delivery of Tapentadol Hydro- chloride. Comput. Math. Method M 2013, 2013, 625729.
-
- 28. El-Zahaby, S. A.; Kassem, A. A.; El-Kamel, A. H. Design and Evaluation of Gastroretentive Levofloxacin Floating Mini-Tablets-In-Capsule System for Eradication of Helicobacter pylori. Saudi. Pharm. J. 2014, 22, 570-579.
-
- 29. Ek, M.; the European Pharmacopoeia, 8th Edition; the European Directorate for the Quality of Medicines & HealthCare of the Council of Europe (EDQM): Strasbourg, 2013; pp 357-359.
- 30. Pawar, H. A.; Dhavale, R. Development and Evaluation of Gastroretentive Floating Tablets of an Antidepressant Drug by Thermoplastic Granulation Technique. Beni Suef Univ. J. Basic Appl. Sci. 2014, 3, 122-132.
-
- 31. Costa, P.; Manuel, J.; Lobo, S. Modeling and Comparison of Dissolution Profiles. Eur. J. Pharm. Sci. 2001, 13, 123-133.
-
- 32. Sreedharan, S. M.; Singh, R. Ciprofloxacin Functionalized Biogenic Gold Nanoflowers as Nanoantibiotics Against Pathogenic Bacterial Strains. Int. J. Nanomed. 2019, 14, 9905-9916.
-
- 33. Vo, A. Q.; Feng, X.; Morott, J. T.; Pimparade, M. B.; Tiwari, R. V.; Zhang, F.; Repka, M. A. A Novel Floating Controlled Release Drug Delivery System Prepared by Hot-Melt Extrusion. Eur. J. Pharm. Biopharm. 2016, 98, 108-121.
-
- 34. Koca, M.; Özakar, E.; Sevinç Özakar, R. Triiyodoanilin’in Sentezlenmesi, Nanosüspansiyonlarının Hazırlanması, İn Vitro Karakterizasyonu ve Radyokontrast Özelliklerinin İncelenmesi. J. Fac. Pharm. Ankara 2021, 45, 264-283.
-
- 35. Gulen, B.; Demircivi, P. Synthesis and Characterization of Montmorillonite/ciprofloxacin/TiO2 Porous Structure for Controlled Drug Release of Ciprofloxacin Tablet with Oral Administration. Appl. Clay Sci. 2020, 197.
-
- 36. Shoukri, R. A.; Ahmed, I. S.; Shamma, R. N. In Vitro and In Vivo Evaluation of Nimesulide Lyophilized Orally Disintegrating Tablets. Eur. J. Pharm. Biopharm. 2009, 73, 162-171.
-
- 37. Rahamathulla, M.; Hani, U.; Alqahtani, A.; Gangadharappa, H. V.; Begum, M. Y.; Jaffer, M.; Osmani, R. A. M.; Chidambaram, K.; Moin, A.; Shankar, S. J. 23 Factorial Design and Optimization of Effervescent Floating Matrix Tablet of Neratinib. J. Pharm. Innov. [Online early access]. DOI: 10.1007/s12247-021-09569-y. Published Online: June 1, 2021. https://doi.org/10.1007/s12247-021-09563-4 (accessed Jan 28, 2022)
-
- 38. Williams, B. B.; Gidley, J. L.; Guin, J. A.; Schechter, R. S. Characterization of Liquid-Solid Reactions. Hydrochloric Acid-Calcium Carbonate Reaction. Ind. Eng. Chem. Fundamen. 1970, 9, 589-596.
-
- 39. Berardi, A.; Bauhuber, S.; Sawafta, O.; Warnke, G. Alginates as Tablet Disintegrants: Understanding Disintegration Mechanisms and Defining Ranges of Applications. Int. J. Pharmaceut. 2021, 601.
-
- 40. Del Gaudio, P.; Colombo, P.; Colombo, G.; Russo, P.; Sonvico, F. Mechanisms of Formation and Disintegration of Alginate Beads Obtained by Prilling. Int. J. Pharmaceut. 2005, 302, 1-9.
-
- 41. Mandal, S.; Basu, S. K.; Sa, B. Sustained Release of a Water-Soluble Drug from Alginate Matrix Tablets Prepared by Wet Granulation Method. AAPS Pharmscitech 2009, 10, 1348-1356.
-
- 42. Jin, S. G.; Yousaf, A. M.; Kim, K. S.; Kim, D. W.; Kim, D. S.; Kim, J. K.; Yong, C. S.; Youn, Y. S.; Kim, J. O.; Choi, H. G. Influence of Hydrophilic Polymers on Functional Properties and Wound Healing Efficacy of Hydrocolloid Based Wound Dressings. Int. J. Pharmaceut. 2016, 501, 160-166.
-
- 43. Hou, L.; Wu, P. Exploring the Hydrogen-bond Structures in Sodium Alginate Through Two-Dimensional Correlation Infrared Spectroscopy. Carbohydr. Polym. 2019,205, 420-426.
-
- 44. Ciolacu, D. E. Structure-Property Relationships in Cellulose-Based Hydrogels. In Cellulose-Based Superabsorbent Hydrogels; Mondal, I. H., Ed.; Springer: Cham, 2018; pp 1-32.
- 45. Raza, A.; Shen, N.; Li, J. H.; Chen, Y. S.; Wang, J. Y. Formulation of Zein Based Compression Coated Floating Tablets for Enhanced Gastric Retention and Tunable Drug Release. Eur. J. Pharm. Sci. 2019, 132, 163-173.
-
- 46. Arza, R. A. K.; Gonugunta, C. S. R.; Veerareddy, P. R. Formulation and Evaluation of Swellable and Floating Gastroretentive Cipro- floxacin Hydrochloride Tablets. AAPS Pharmscitech 2009, 10, 220-226.
-
- 47. Wongsasulak, S.; Puttipaiboon, N.; Yoovidhya, T. Fabrication, Gastromucoadhesivity, Swelling, and Degradation of Zein-Chitosan Composite Ultrafine Fibers. J. Food Sci. 2013, 78, N926-N935.
-
- 48. Vanitha, K.; Varma, M.; Ramesh, A. Floating Tablets of Hydralazine Hydrochloride: Optimization and Evaluation. Braz J. Pharm. Sci. 2013, 49, 811-819.
-
- 49. Shinde, A. J.; Patil, M. S.; More, H. N. Formulation and Evaluation of an Oral Floating Tablet of Cephalexin. Indian J. Pharm. Educ. 2010, 44, 243-252.
- 50. Raza, A.; Bukhari, N. I.; Karim, S.; Hafiz, M. A.; Hayat, U. Floating Tablets of Minocycline Hydrochloride: Formulation, In-Vitro Evaluation and Optimization. Futur. J. Pharm. Sci. 2017, 3, 131-139.
-
- 51. Patel, V. R.; Amiji, M. M. pH-Sensitive Swelling and Drug-Release Properties of Chitosan Poly(ethylene oxide) Semi-Interpenetrating Polymer Network. Acs Sym. Ser. 1996, 627, 209-220.
-
- 52. Marchais, H.; Cayzeele, G.; Legendre, J. Y.; Skiba, M.; Arnaud, P. Cross-linking of Hard Gelatin Carbamazepine Capsules: Effect of Dissolution Conditions on In Vitro Drug Release. Eur. J. Pharm. Sci. 2003, 19, 129-132.
-
- 53. Abou Youssef, N. A. H.; Kassem, A. A.; El-Massik, M. A. E.; Boraie, N. A. Development of Gastroretentive Metronidazole Floating Raft System for Targeting Helicobacter pylori. Int. J. Pharmaceut. 2015, 486, 297-305.
-
- 54. Young, C. R.; Dietzsch, C.; McGinity, J. W. Compression of Controlled-Release Pellets Produced by a Hot-Melt Extrusion and Spheronization Process. Pharm. Dev. Technol. 2005, 10, 133-139.
-
- 55. Bizarro, M.; Rodil, S. E. Physicochemical Characterization of Photocatalytic Materials. In Photocatalytic Semiconductors: Synthesis,Characterization, and Environmental Applications, Medina-Ramírez, A.; Hernández.-Ramírez, A. Ed. Springer: Cham, 2015; p 127.
-
- 56. Gandolpho Tótoli, E.; Regina Nunes Salgado, H. Development and Validation of the Quantitative Analysis of Ampicillin Sodium in Powder for Injection by Fourier-transform Infrared Spectroscopy (FTIR). Physical Chemistry 2013, 2, 103-108.
-
- 57. Queiroz, A. C.; Santos, J. D.; Monteiro, F. J. Development of a System to Adsorb Drugs Onto Calcium Phosphate Materials. J. Mater Sci. Mater Med. 2005, 16, 641-646.
-
- 58. Murei, A.; Ayinde, W. B.; Gitari, M. W.; Samie, A. Functionalization and Antimicrobial Evaluation of Ampicillin, Penicillin and Vancomycin with Pyrenacantha grandiflora Baill and Silver Nanoparticles. Sci. Rep. 2020, 10, 11596.
-
- 59. Rogowska, A.; Rafinska, K.; Pomastowski, P.; Walczak, J.; Railean-Plugaru, V.; Buszewska-Forajta, M.; Buszewski, B. Silver Nanoparticles Functionalized with Ampicillin. Electrophoresis 2017, 38, 2757-2764.
-
- 60. Awasthi, R.; Kulkarni, G. T.; Pawar, V. K.; Garg, G. Optimization Studies on Gastroretentive Floating System Using Response Surface Methodology. AAPS Pharmscitech 2012, 13, 85-93.
-
- 61. Bajpai, S. K.; Shah, F. F.; Bajpai, M.; Jadaun, M.; Jyotishi, P. Dynamic Release of Amoxicillin from Orally Dissolving Film (ODF) Composed of Casein and Sodium alginate. J. Drug. Res. Dev. 2017, 3, DOI: 10.16966/2470-1009.134.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2022; 46(2): 145-158
Published online Mar 25, 2022
- 10.7317/pk.2022.46.2.145
- Received on Oct 5, 2021
- Revised on Nov 24, 2021
- Accepted on Dec 19, 2021
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
- Conflict of Interest
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Rukiye Sevinç Özakar, and Emrah Özakar
-
Department of Pharmaceutical Technology, Faculty of Pharmacy, Atatürk University, Erzurum, Turkey
- E-mail: emrahozakar@atauni.edu.tr
- ORCID:
0000-0002-2972-8084, 0000-0002-7443-208X
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr