






- Curing Study of the New Silane Monomers and Their IPNs
Firas Jameel Jabbar†

Department of Pharmacy, Southern Technical University, Basrah, Iraq
- 새로운 Silane 모노머와 IPN 중합체의 경화 연구
Reproduction, stored in a retrieval system, or transmitted in any form of any part of this publication is permitted only by written permission from the Polymer Society of Korea.
Interpenetrating polymer networks (IPNs) that have silane resins as one of their components are industrially important due to their physical and chemical properties. The current study included preparation and characterization of two types of unsaturated silane monomers in order to prepare new types of IPNs with unsaturated polyesters (palatal). These monomers were characterized with Fourier transform infrared spectroscopy (FTIR) and nuclear magnetic resonance spectroscopy (1H NMR), and their molecular weights were determined by cryoscopy technique. The curing properties of the silane monomers and the IPNs were observed by differential scanning calorimetry (DSC), and several of thermal curing functions were determined, such as the initial curing temperature, optimum curing temperature, curing energy, activation energy and rate of curing. There was a significant decrease in the initial and maximum curing temperatures compared with the unsaturated polyester resin (palatal) due to the decrease in the activation energy from 105.4 to 77.3 ºC when the allyoxy silane monomer percentage was shifted from 5 to 20%. Meanwhile, the activation energy for the IPNs based on α-methyl-butenoxy silane decreased from 96.8 to 55.8 kJ/mol.
Two types of unsaturated silane monomers were prepared and characterized for the purpose of using them in the preparation of new types of interpenetrating polymer networks (IPNs) with unsaturated polyesters (Palatal). Several thermal curing functions were determined, such as the initial curing temperature, optimum curing temperature, curing energy, activation energy and rate of curing. There is a significant decrease in the initial curing temperatures and maximum curing temperatures compared with the unsaturated polyester resin (Platel).
Keywords: silane monomer, interpenetrating polymer networks, curing, differential scanning calorimetry, palatal, cryoscopy.
An interpenetrating polymer network (IPN) comprises two polymeric networks, one of which must at least have been synthesized and/or crosslinked in the presence of the other.1-3 However, there is no chemical linkage between these types of distinct networks.4,5 Preparing IPNs is often distinct from other methods that mix two or more polymers, such as physical blends or copolymerization.6-8 In certain cases, interpenetrating polymer networks (IPNs) can be made by polymerizing free radicals or cations with two multifunctional monomers or telechelic oligomers.9-11 Several types of IPNs exist, with each being influenced by the method used to synthesize multicomponent alloy.12-14 An IPN can be prepared by the sequential technique, which is by swelling a crosslinked polymer in a monomer and curing the agent of its corresponding polymer with subsequent polymerization of the monomer in swollen polymer matrix.15,16 When IPN is in the form of latex, it is known as latex IPN; it is also known as interpenetrating elastomeric networks (IENs).17,18
Another method of synthesizing IPNs is the simultaneous (SIN) technique. This technique requires blending the linear polymers, prepolymers, or monomers in some liquid from latex, solution or bulk together with their respective crosslinking agent, followed by evaporating the liquid vehicle, if present, and simultaneously curing the component polymers.19,20 If only one polymer of the combined polymers is crosslinked, then the material is referred to as semi-IPNs.21-23 There are a large number of polymers that have the ability to form lattice structures between them, and they are bonded with physical bonds (van der Waals) to produce constitutive networks, resulting in a broad range of properties that have different applications, including, in biomedical specialties, separation membranes and selective absorbents.24,25
The importance of IPNs lies in their ability to produce polymers with improved properties as a result of combining the unique properties of the polymers that form the IPNs. For example, it is possible to produce IPNs that possess the characteristic toughness and high thermal stability from mixing two incompatible polymers, thermoplastics and thermosets.26-28 Lin et al.29 were able to produce a new type of IPNs that included unsaturated polyesters with resoling, a type of phenolic resin. These polymers have a unique property of thermal stability in addition to their ability to prevent the formation of smoke and toxic gases during combustion.
Guhanathan et al.30 were able to prepare a three-component system of IPNs, including polyethylene-acrylonitrile-unsaturated polyurethane. This type of polymer possesses the mechanical properties (flexural and impact strength) surpassing that of unsaturated polyesters alone. Chung et al.31 were able to produce new types of hybrid IPNs that contain inorganic compounds in their composites. They reached the gelatinous state of alkoxysilane at a temperature of 60 ºC by using the nuclear magnetic spectroscopy technique. It was observed that the unsaturated polyester resin decomposed upon treatment with HCl acid. Finally, they “were able to form the IPNs by using the photopolymerization method of unsaturated polyesters resin by forming π-interactions between the aromatic ring of the unsaturated polyester resin and phenyltrimethoxysilane.
New IPNs have been referred to in recent years, and they are used in a variety of medical applications (hydrogels IPNs). They are divided into categories based on how the polymers are mixed or prepared. The first is known as a mechanical blend (no chemical bonds between the polymers) and the second is known as a graft copolymer (containing primary bonds between the polymeric components).32-34 Tissue engineers have long attempted to replicate native tissue's hydrophilicity and hierarchical architecture. These two properties work together to enable large-scale molecular rearrangements of the constituent bioactive polymers, resulting in specific material properties.35-37 Mahou et al.38 created a hydrogel IPN matrix containing collagen-quinine. These natural polymers showed great resistance to enzymes without altering the property of the embedded mesenchymal stromal cells. Compared to collagen, the results of the study showed a promising application of the IPN microspheres in the field of tissue engineering. Soni et al.39 investigated a pH-dependent system that included Poly(alcohol-carboxylate pullulan) loaded with the drug pirfenidone-loaded IPN microsphere. The results showed that the polymer degradation process in the stomach and intestine was needed to control the value of the pH. In addition to that, during a laboratory study of the enzyme degradation by pullulanase, it was observed that there were no toxic complications for the IPNs.
The present paper was focused on the synthesis of IPNs based on Palatal-silicone as well as investigating the curing parameters by using differential scanning calorimetry (DSC) technique.
Materials and Instruments. In this study, high purity chemicals were used. Silicon tetrachloride and allyl alcohol were supplied by H & W L.T.D (England). Sodium hydrogen carbonate, anhydrous magnesium sulphate, ethyl methyl ketone, peroxide, cobalt accelerator and unsaturated polyester (Palatal-P50T) were supplied by Al-Basil Applied research Center. Silicon tetra chloride, allyl alcohol and 4-penten-2-ol were obtained from Fluka A.G. (Chemische, Fabrika).
The curing kinetics of IPNs were measured using a NETZSCH 204F1 differential scanning calorimetry (DSC, NETZSCH, Bavarian, Germany). The specimens were heated under a flow of nitrogen (50 mL/min-1) from 20 to 350 ºC at a heating rate of 5, 10, 15 and 20 ºC/min, respectively. On a Perkin-Elmer 16F PC FTIR spectrometer (Perkin Elmer Cetus, Norwalk, CT), the IR spectrum was registered. ECX 100-JEOL HNMR spectrometer using CDCl3 as a solvent.
The FEI Q3D scanning electron microscope (SEM, Zeiss, Germany) was used to obtain the micrographs of the samples. Aknauer-cryoscopic unit No.2400 was used to determine the number of average molecular weight (Mn). The instrument was calibrated with benzyl solution of suitable molal concentration.
Preparation of the Tetra Allyloxy Silane Monomer and Tetra (α-methyl butenoxy) Silane. To prepare the monomer (tetra allyloxy silane), the reaction vessel provided with an electrical mixer was charged by 28.33 gm (0.166 mol) of silicon tetra chloride and 36.66 gm (0.666 mol) of allyl alcohol, and the reaction was maintained for three hours at a temperature of 50 ºC. Upon the completion of the reaction, the hydrogen chloride formed as a byproduct was neutralized by sodium hydrogen carbonate. Then, the product was distilled under vacuum pressure to obtain a pale-yellow oily liquid, and characterized by using FTIR and 1H NMR spectroscopy. Its average molecular weight was determined by a cryoscopic instrument. The results proved the correct expected chemical structure.
As for the preparation of the monomer tetra (α-methyl- butenoxy) silane, the following quantities of 28.33 gm (0.166 mol) of silicon tetra chloride and 57.33 gm (0.667 mol) of 4-penten-2-ol were used. Then, the same procedure for preparing the monomer tetra allyoxy silane was followed. The product was also in the form of a yellowish oily liquid. It was characterized by FTIR and 1H NMR, and its average molecular weight was determined via cryoscopic instrument. The results proved the correct expected structure.
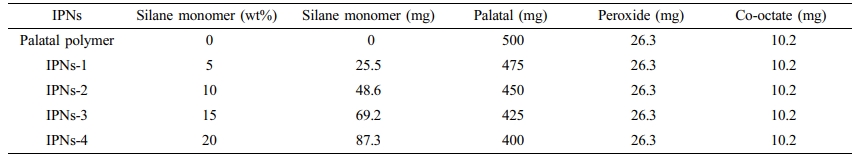
Preparation of the IPNs Based on (Silane Monomer-Palatal). Several IPNs consisting different weight percentages of the silane monomers and Palatal were prepared by efficiently mixing the predetermined quantities of the two components as shown in Table 1. The silane monomer and Palatal were mixed gently for about 2 min at room temperature. Then, ethyl methyl ketone peroxide and cobalt octate accelerator were added, and mixed gently. Samples from the obtained product were taken for DSC curing study and the residue was left overnight at room temperature.
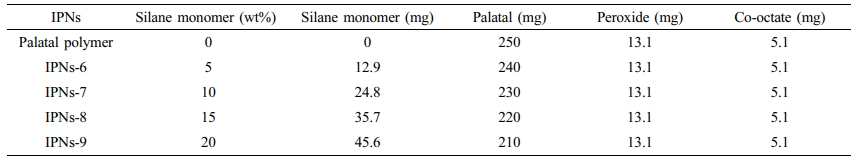
Preparation of the IPNs Based on (Silane-Palatal). Several IPNs consisting of different weight percentages of the silane monomers and Palatal were prepared by efficiently mixing the predetermined quantities of the two components as shown in Table 2.
The tetra (α-methyl butenoxy) silane and Palatal were mixed gently for about 2 min at room temperature. Then, ethyl methyl ketone peroxide and cobalt octate accelerator were added and mixed gently. Samples from the obtained product were taken for DSC curing study, and the residue was left overnight at room temperature.
|
Table 1 Typical Formula Used for the Preparation of the IPNs Based on Palatal-Allyloxy Silane Monomers |
|
Table 2 Typical Formula Used for the Preparation of the IPNs Based on Palatal-Tetra (α-methyl butenoxy) Silane Monomers |
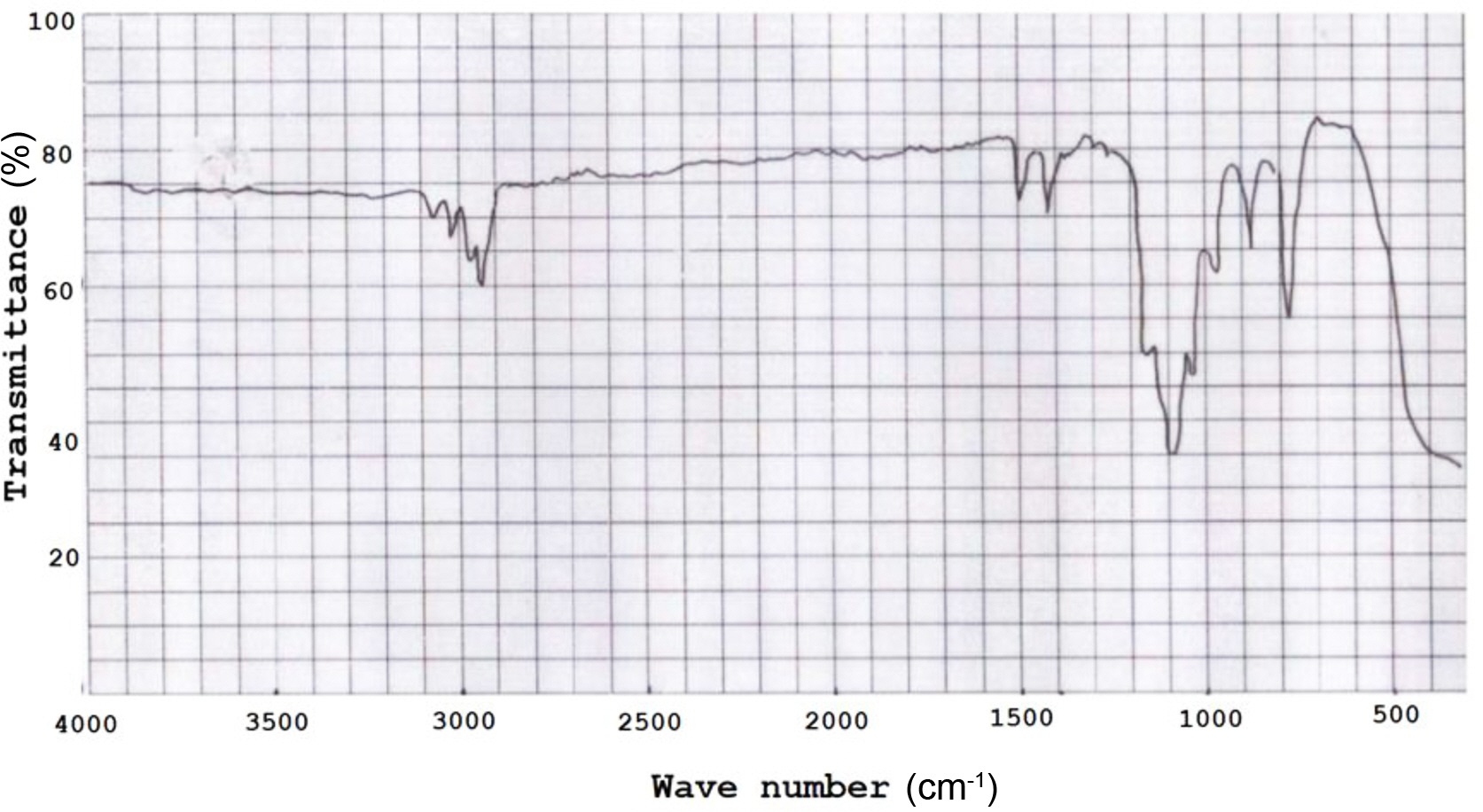
The results of the characterization the tetra allyloxy silane monomer with Fourier transform infrared spectroscopy (FTIR) are shown in Figure 1, where the (4000-400) cm-1 region is exhibited as thin films over NaCl discs. The essential absorption bands observed showed strong broad bands in the region 1050 cm-1 as attributed to the stretching vibration of the Si-OC. Other bands were also observed in the region 850 and 910 cm-1 due to the stretching vibration of the alkoxy group (--OR) where R was the allyl group. There were also two distinguished bands: first was in the region (3020 cm-1) related to the stretching vibration of the olefinic protons and the other was in the region (1620 cm-1) related to the stretching vibration of the C=C of the allyl group.
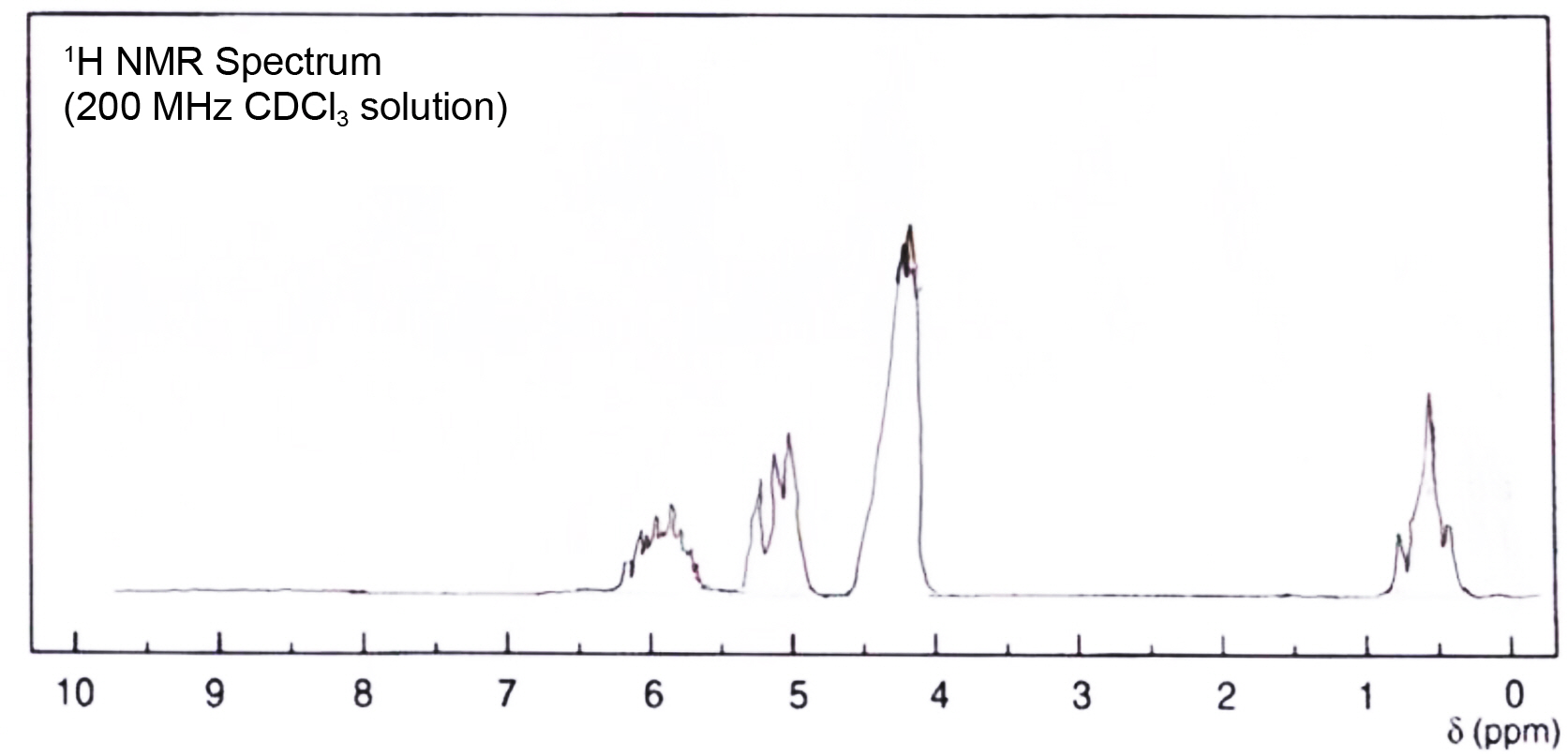
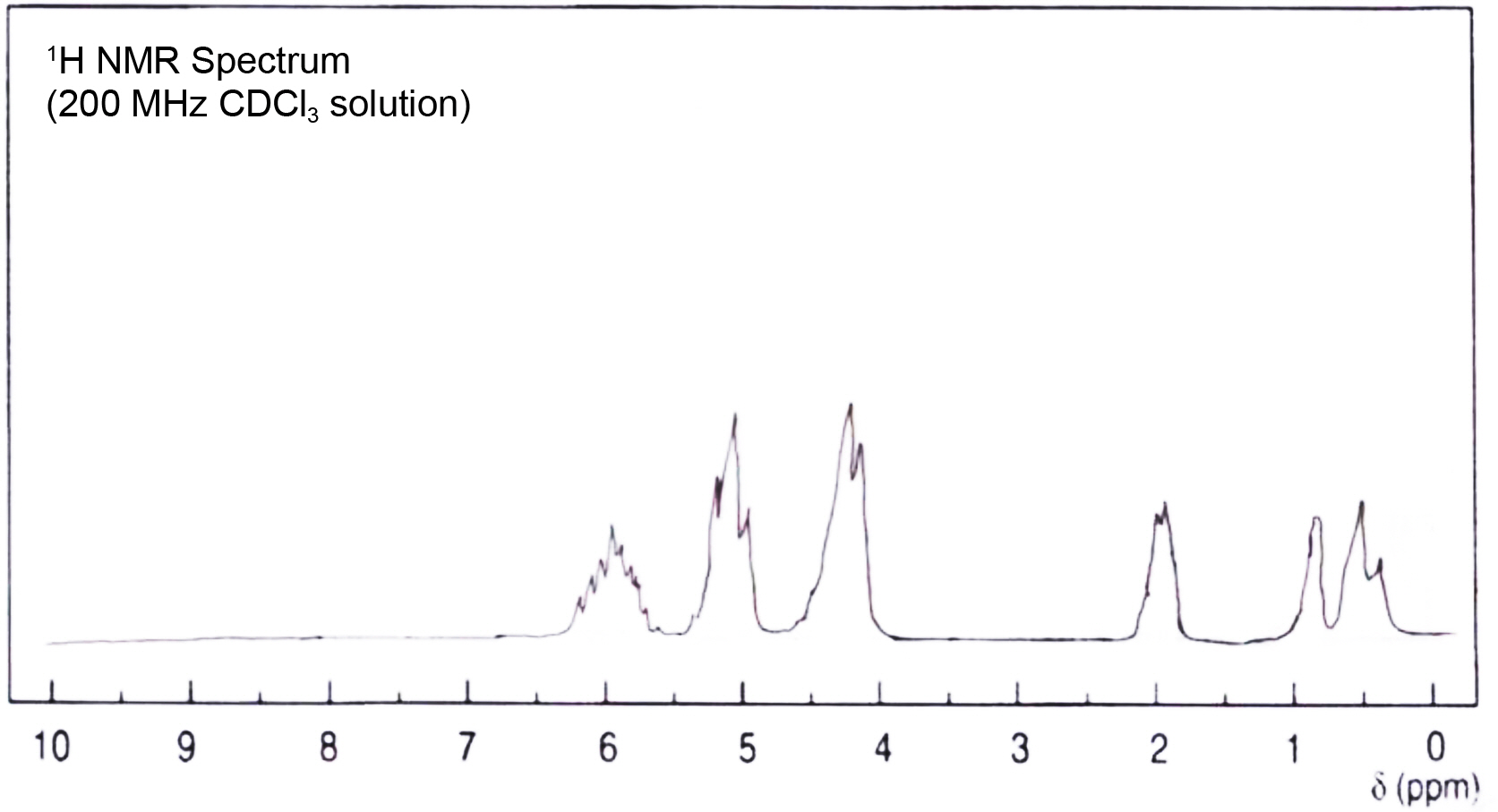
The 1H NMR spectra of the allyloxy silane monomer in Figure 2 indicated the appearance of the allylic protons and disappearance of the signal related to the OH protons of the allyl alcohol in the region δ = 4 ppm. The allylic protons --HC=CH-- of these monomers showed multiple signals in the downfield region δ = 5.7-6.3 ppm. The protons of the methylene group -OCH2- of these monomers showed signals in the region δ = 4.3-4.4 ppm. In the case of the monomer tetra (α-methyl butenoxy) silane, sharp signals in the higher field region δ = 0.2-0.3 ppm were observed and related to the Si(CH3)2 and -Si(CH3), respectively. The protons of the methylene group CH-CH2-CH- showed signals in the region δ = 2 ppm and the protons of methyl group -OCHCH3 were absorbed in region δ = 1.05 ppm. The average molecular weight of both silane monomers was determined by freezing point depression measurement (F.D) using Knauer cryoscopic unit. The average molecular weight of tetra allyoxy silane was 252±6 while the calculated value was 256. The average molecular weight of tetra (α-methyl butenoxy) silane was 360±4 while the calculated value was 368.
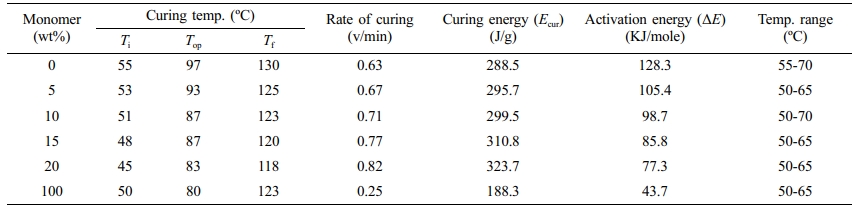
The curing thermograms were analyzed and several curing parameters were determined as listed in Tables 3 and 4.
a) Initial curing temperature (Ti): which was determined from the thermograms as a deflecting point of the curve from tangent to the base line.
b) Optimum curing temperature (TOP): Which was taken as the temperature when the rate of curing reaches optimum.
c) Final curing temperature (Tf): which was taken as the temperature at which the curing reactions ars completed and the curve returns to the baseline.
d) Curing energy (Ecur): which was calculated from the area under the curve relatively to the standard indium melting peaks.
e) The rate curing (d(ΔH)/dt: was measured at different temperatures from instantaneous slops of the DSC curves.
f) The activation energy of the curing reaction (ΔE): determined from the DSC thermograms, by plotting the rate of curing at different temperatures at the initial stages of the curing process < 15% against 1/T.
g) The ΔE was determined from the slope of the Arrhenious plotes:

Where R is the gas constant = 8.314 J·mol-1 K-1.
The curing of these monomers was initiated by ethyl methyl ketone peroxide and cobalt octate as accelerators. The curing reaction took place according to the free radical mechanism at the double bond to produce silicone resin, as shown in the following expected equations:
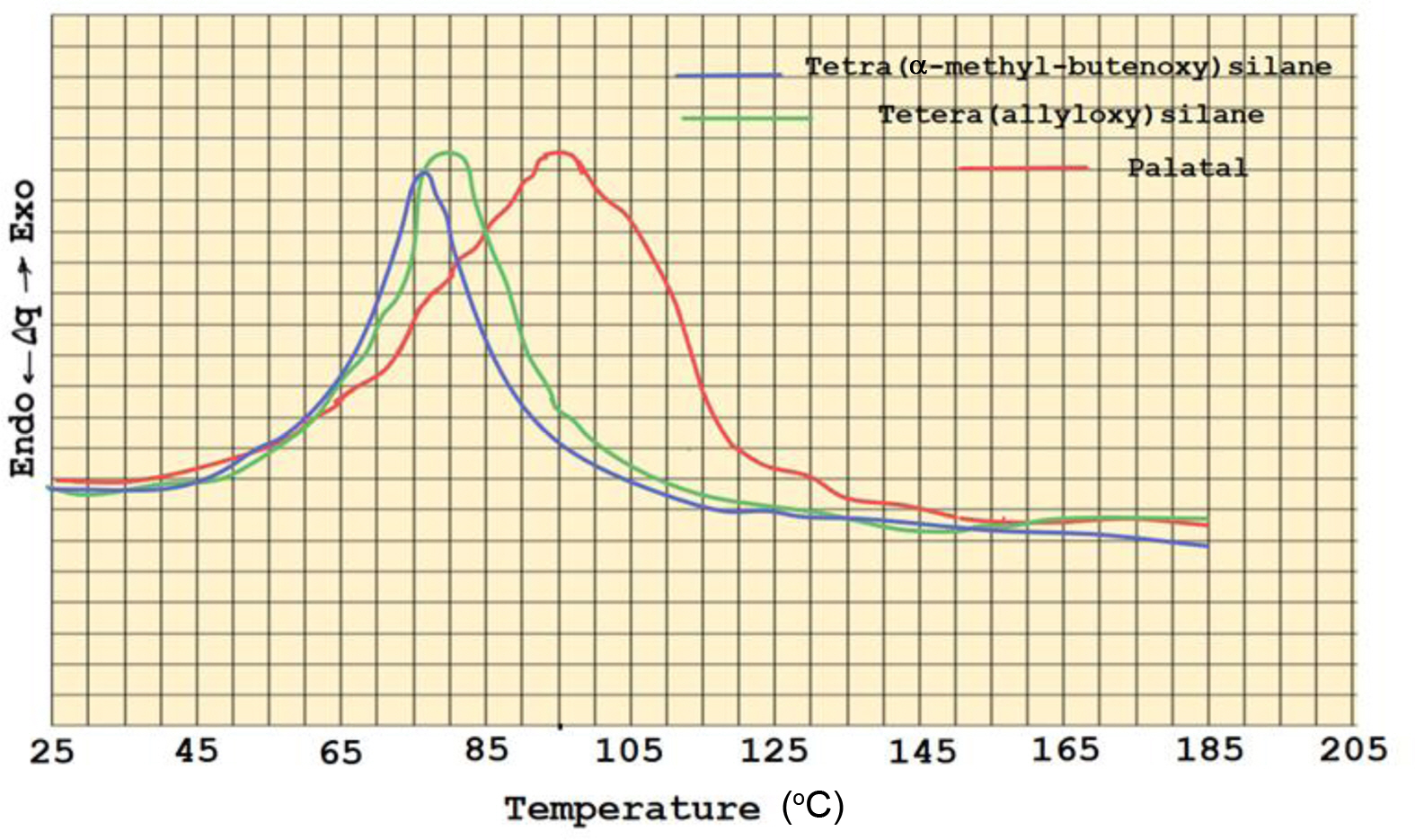
Similar curing behavior was valid with tetra (α-methyl butenoxy) silane. Figure 7 shows the DSC thermograms of tetra (allyoxy) silane and tetra (α-methyl butenoxy) silane monomers, indicating the presence of exothermic curing peak that has an optimum curing temperature at 80 and 77 ºC with activation energies 43.7 and 33.5 kJ/mol for two monomers, respectively.
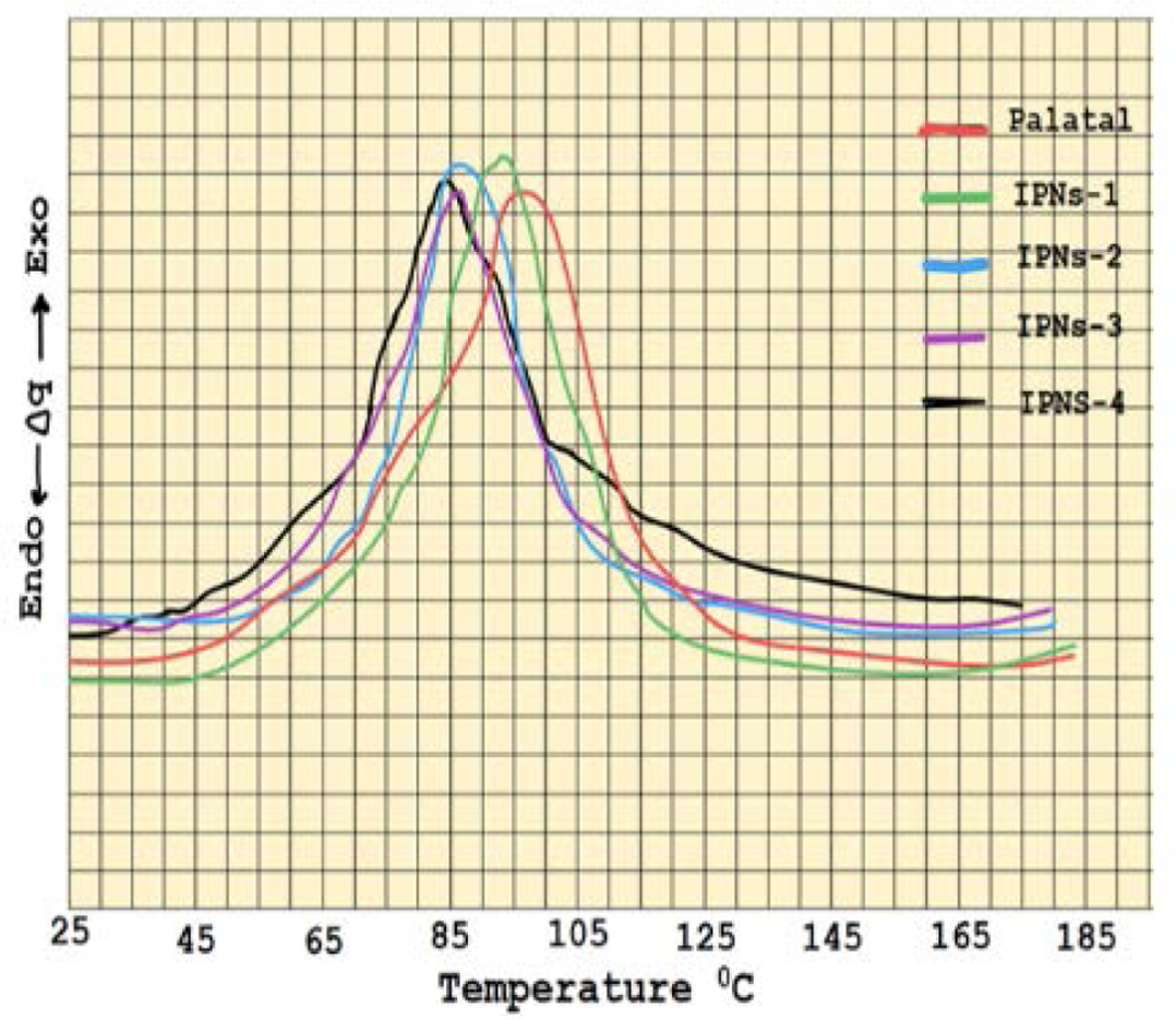
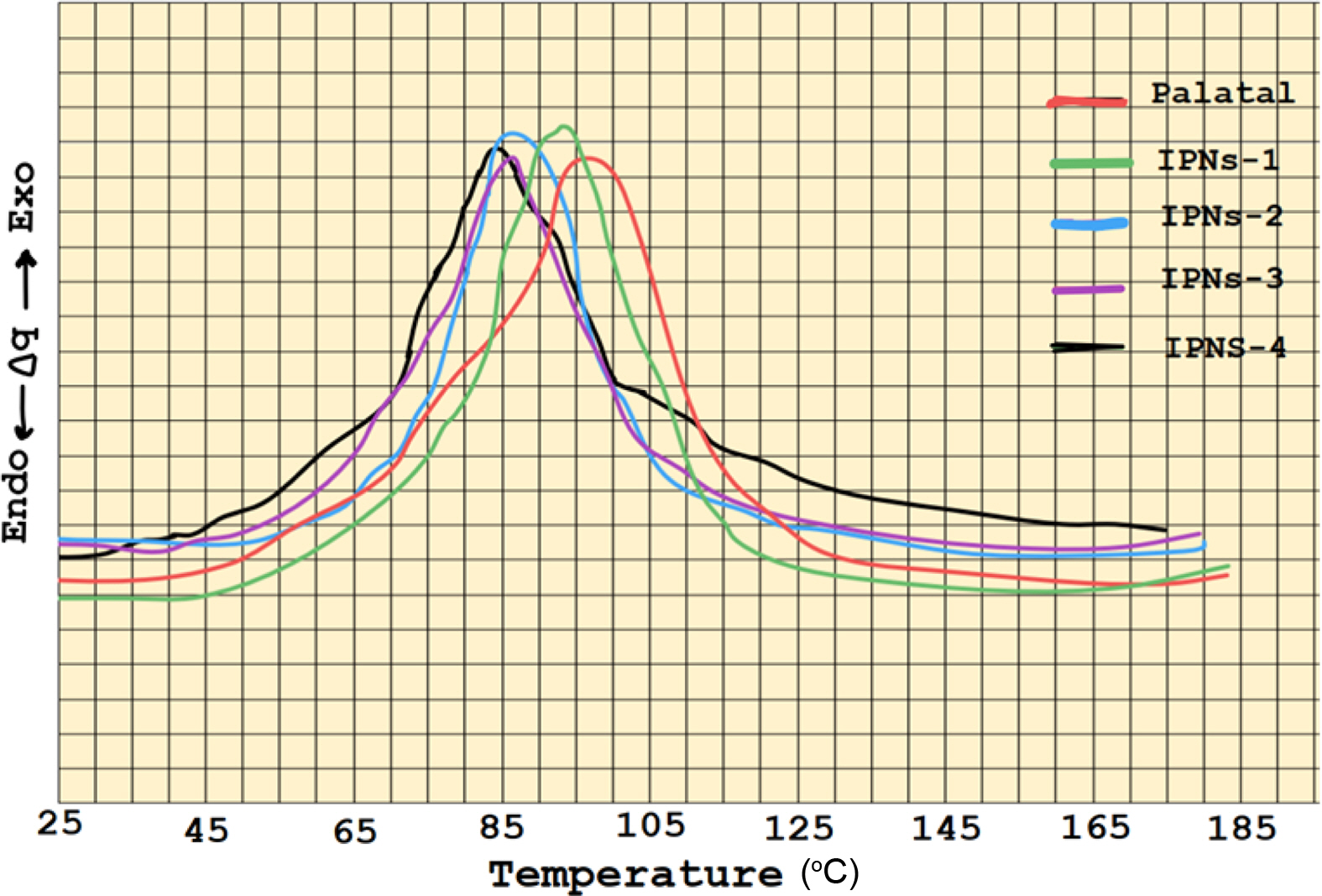
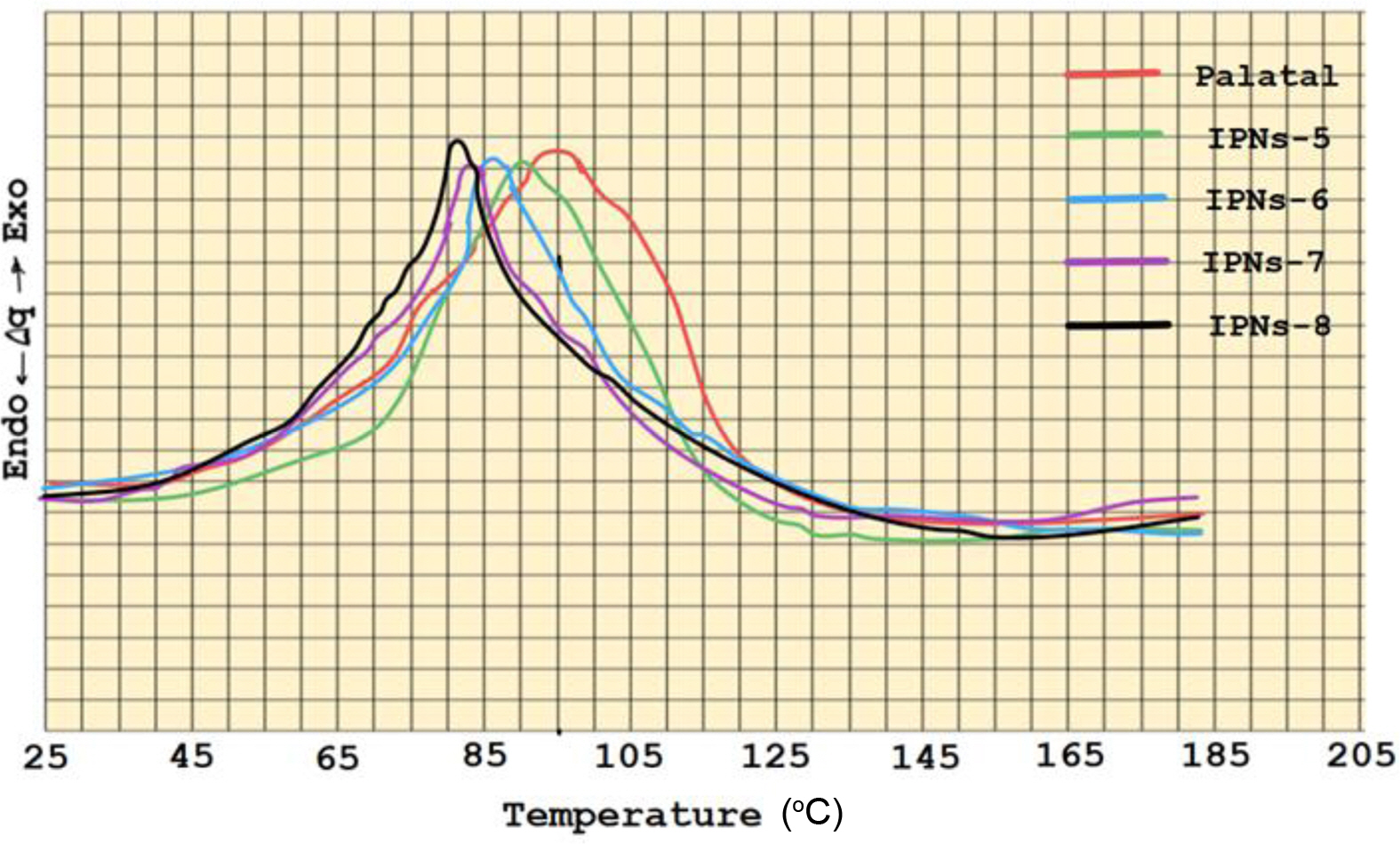
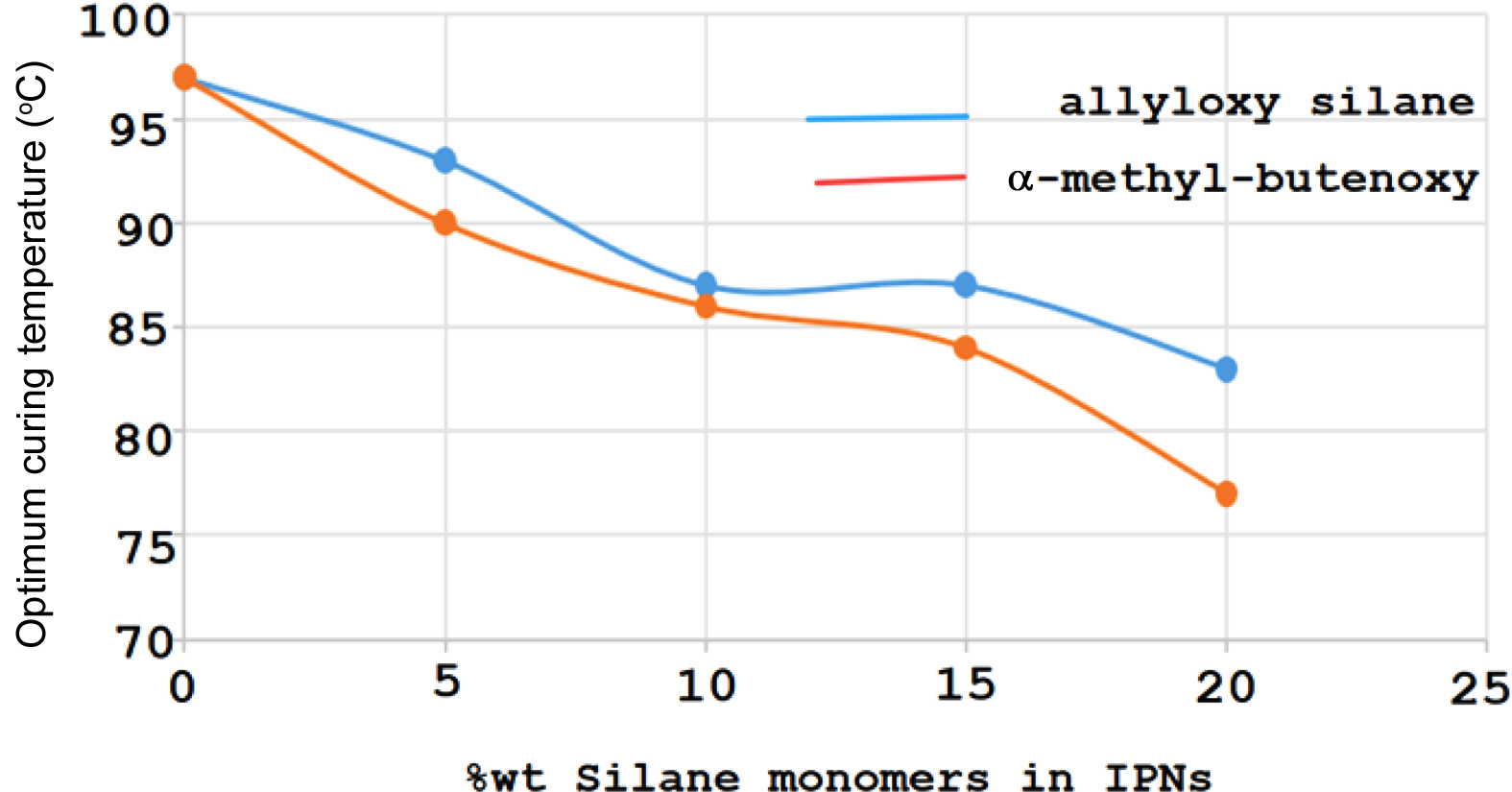
The IPNs curing results of allyoxy and α-methyl butenoxy silane monomers with Palatal resin which are prepared in different weight percentages 5-20% of the silane monomers are shown in Tables 1 and 2. The DSC thermograms for the IPNs of both silane monomers (Figures 5 and 6) with Palatal showed one exothermic curing peak, confirming the formation of simultaneous IPNs. In addition to that, the DSC thermograms indicated that the exothermic curing peak of Palatal at the optimum curing temperature (97 ºC) shifted to higher curing temperature with increasing percentages of the reactive silane monomers. For example, in the case of both monomers, tetra allyloxy silane and tetra (α-methyl butenoxy) silane, the peak for curing temperature shifted from 97 ºC for Palatal resin to 83 ºC and 81 ºC for the IPNs containing 20% of monomers, respectively. Therefore, these silane monomers increased the reactivity of the Palatal towards polymerization which was very important from the technological point of view.
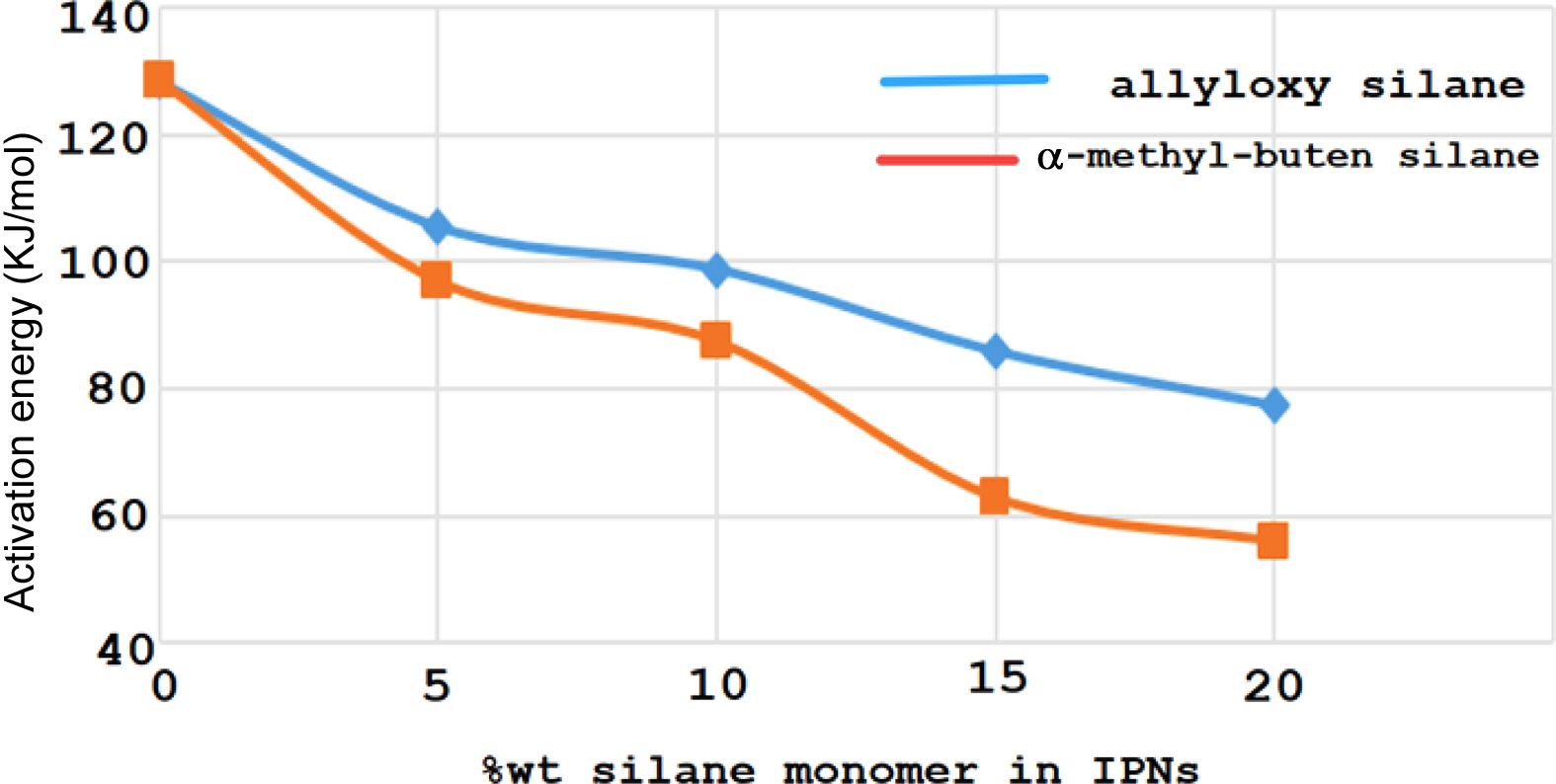
The rate of curing also increased with increasing weight percentages of the silane monomers (Tables 2 and 3). From the results shown in Tables 1 and 2, the monomer tetra (α-methyl butenoxy) silane had better cure properties than monomer tetra(allyloxy) silane in its ability to form IPNs with palatal resin as evident in Figure 7. The relationship between the monomer ratio in the compositions of IPN5-8 showed that there was a significant decrease in the activation energy value when the percentage of monomers increased. Moreover, from Figure 8, there was an important reduction in the curing temperature of the IPNs that had tetra (α-methyl butenoxy) silane in their composition compared to the IPNs that contained the tetra allyloxy silane.
The results of the thermal curing technique of the IPNs could be compared using the DSC device as shown in Table 5. The activation energy of the IPNs consisting of palatal and (tetra allyloxy silane) significantly decreased by increasing the ratio of monomer (tetra allyloxy silane) from 128.3 to 77.3 kJ/mol.
The same effect was observed for the monomer tetra (a-methyl butenoxy) silane, with a decrease from 128.5 to 55.8 kJ/mol. However, the effect of monomer tetra (a-methyl butenoxy) silane was greater than that of monomer (tetra allyloxy silane), lowering the value of the IPNs’ activation energy. This change can be attributed to the effect of the (a-CH3) group in the monomer (a-methyl butenoxy); with the former pushing the polymeric chains from each other or in polymeric network. In addition to that, from Table 5, there was a significant increase in the curing energy needed for the process of polymerization which formed IPNs that included tetra (a-methyl butenoxy) silane with palatal compared with the IPNs that included tetra allyloxy silane and palatal. This can be attributed to the effect of the electron-donating group (a-CH3) on the rate of curing.
There is a lack of studies related to the preparation of IPNs that include silane monomers palatal. However, some references have been obtained, including the preparation of IPNs consisting of silane monomers with epoxy resin.40-42 These studies have confirmed that these monomers have a similar effect to the ones obtained in this study with regard to decreasing the activation energy and increasing the curing energy.43-45
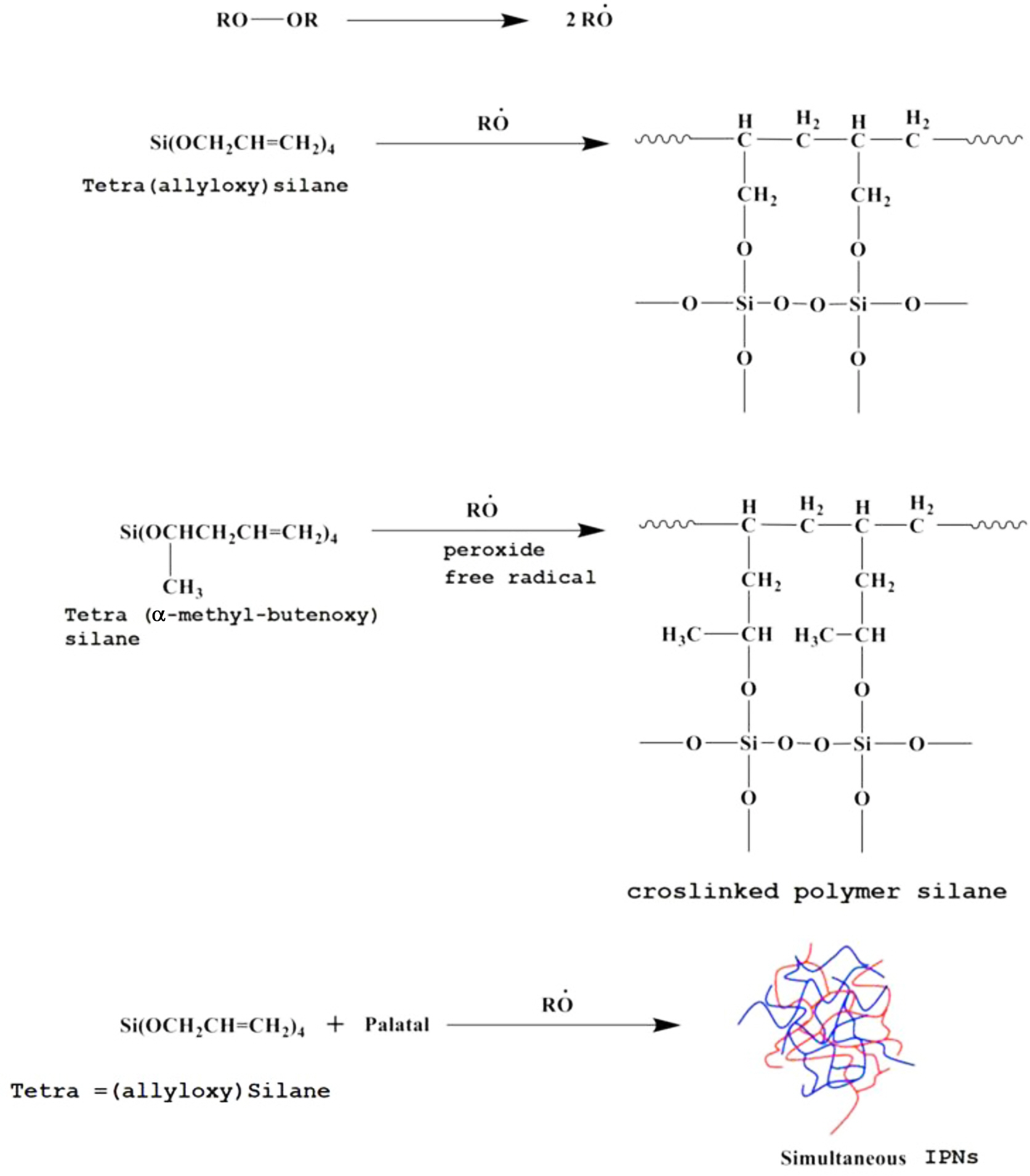
Scheme 1. IPN formation
Figure 3, 4, 9
|
Figure 1 FTIR spectrum of terta allyloxy silane monomer. |
|
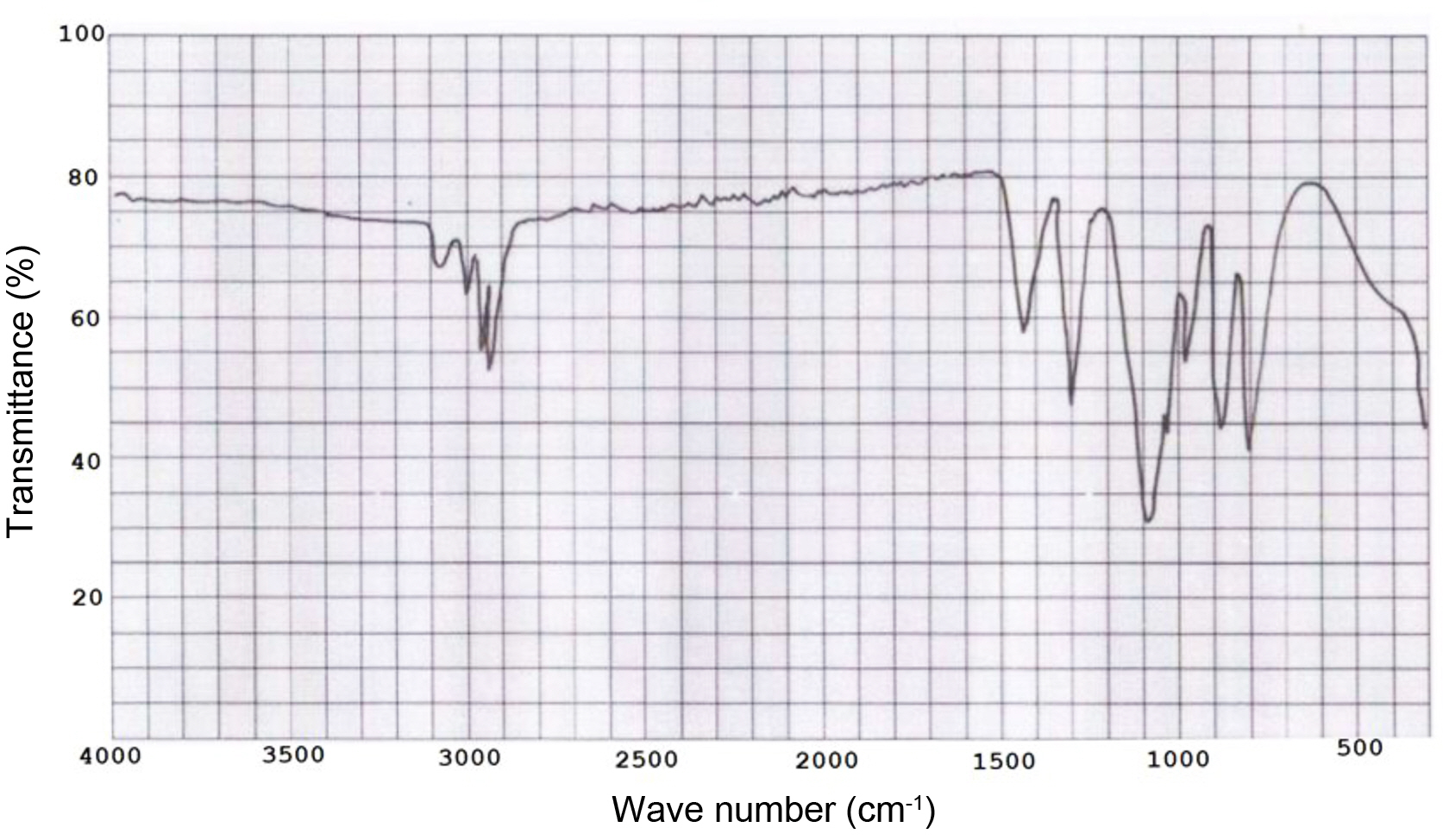
Figure 2 FTIR spectrum of tetra (α-methyl butenoxy) silane monomer |
|
Figure 7 DSC thermograms for palatal-tetra (α-methyl butenoxy) silane and Terta allyloxy silane monomers. |
|
Figure 5 DSC thermograms for palatal-tetra allyloxy silane IPNS of various wt% compositions. |
|
Figure 6 DSC thermograms for palatal-tetra allyloxy silane IPNS of various wt% compositions. |
|
Figure 8 The relationship between wt% of silane monomers and activation energy. |
|
Figure 3 1 H NMR spectrum of terta allyloxy silane monomer. |
|
Figure 4 1 H NMR spectrum of tetra (α-methyl butenoxy) silane monomer. |
|
Figure 9 The relationship between wt% of silane monomers and optimum curing temperature oC. |
|
Table 3 The 1 H NMR Expected Resonance of the Functional Groups of the Silane Monomers |

|
Table 4 DSC Curing Parameters of the IPNs Based on Palatal-Tetra Allyloxy Silane |
|
Table 5 DSC Curing Parameters of the IPNs Based on Palatal-Tetra (α-methyl butenoxy) Silane |
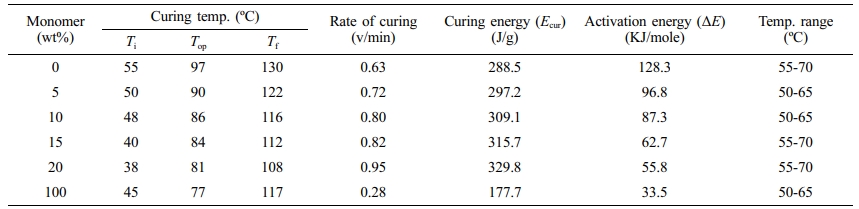
The incorporation of silane monomers into Palatal to prepare the IPNs has improved the curing parameters. The curing temperature of IPNs showed an exothermic curing peak, confirming the formation of simultaneous IPNs while the DSC thermograms indicated that the exothermic curing peak of Palatal at the optimum curing temperature (97 ºC) shifted to higher curing temperature with increasing percentages of the reactive silane monomers. For example, in the case of both monomers tetra allyloxy silane and tetra (α-methyl butenoxy) silane, the peak for curing temperature shifted from 97 ºC for Palatal resin to 83 and 81 ºC for the IPNs containing 20% of monomers, respectively. Since these silane monomers increased the reactivity of the Palatal towards polymerization, the technological implication becomes critical.
- 1. Polymer Blends Handbook; Utracki, L. A., Wilkie, C. A., Eds. Kluwer Academic Publishers: Dordrecht, 2002.
-
- 2. Sperling, L. H.; Mishra, V. B. A. T. The Current Status of Interpenetrating Polymer Networks. Polym. Adv. Technol. 1996, 7, 197-208.
-
- 3. Horák, Z.; Fortelný, I.; Kolařík, J.; Hlavatá, D.; Sikora, A. Polymer Blends. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons: New York, 2005; pp 1-59.
-
- 4. Panteli, P. A.; Patrickios, C. S. Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications. Gels 2019, 5, 36.
-
- 5. Kamal, M. Camphor and Poly(Antimonyacrylate) Based Inter- penetrating Polymer Network: Synthesis and Characterization. Indian J. Chem. Technol. 2011, 18, 284-290.
- 6. Shahrousvand, M.; Ghollasi, M.; Zarchi, A. A. K.; Salimi, A. Osteogenic Differentiation of hMSCs on Semi-interpenetrating Polymer Networks of Polyurethane/poly(2‑hydroxyethyl Meth- acrylate)/cellulose Nanowhisker Scaffolds. Int. J. Biol. Macromol. 2019, 138, 262-271.
-
- 7. De Lima, G. G.; Elter, J. K.; Chee, B. S.; Magalhães, W. L. E.; Devine, D. M.; Nugent, M. J.; de Sá, M. J. A Tough and Novel Dual-response PAA/P (NiPAAM-co-PEGDMA) IPN Hydrogels with Ceramics by Photopolymerization for Consolidation of Bone Fragments Following Fracture. Biomed. Mater. 2019, 14, 054101.
-
- 8. Fan, H.; Gong, J. P. Fabrication of Bioinspired Hydrogels: Challenges and Opportunities. Macromolecules 2020, 53, 2769-2782.
-
- 9. Goczkowski, M.; Gobin, M.; Hindié, M.; Agniel, R.; Larreta-Garde, V. Properties of Interpenetrating Polymer Networks Associating Fibrin and silk Fibroin Networks Obtained by a Double Enzymatic Method. Mater. Sci. Eng., C 2019, 104, 109931.
-
- 10. Khan, J.; Alexander, A.; Saraf, S.; Saraf, S. Biomedical Applications of Interpenetrating Polymer Network Gels. In Interpenetrating Polymer Network: Biomedical Applications;Springer: Singapore, 2020; pp 289-312.
-
- 11. Krishnamoorthy, S.; Zhang, Z.; Xu, C. Biofabrication of Three-dimensional Cellular Structures Based on Gelatin Methacrylate-alginate Interpenetrating Network Hydrogel. J. Biomater. Appl. 2019, 33, 1105-1117.
-
- 12. Chen, C. H.; Chen, M. H. Synthesis, Thermal Properties, and Morphology of Blocked Polyurethane/epoxy Full‐interpenetrating Polymer Network. J. Appl. Polym. Sci. 2006, 100, 323-328.
-
- 13. Myung, D.; Waters, D.; Wiseman, M.; Duhamel, P. E.; Noolandi, J.; Ta, C. N.; Frank, C. W. Progress in the Development of Interpenetrating Polymer Network Hydrogels. Polym. Adv. Technol. 2008, 19, 647-657.
-
- 14. Dave, P. N.; Khosla, E. Blends, Interpenetrating Polymer Networks, and Gels of Unsaturated Polyester Resin Polymers with Other Polymers. In Unsaturated Polyester Resins: Fundamentals, Design, Fabrication, and Applications; Thomas, S., Hosur, M., Chirail, C. J., Eds.; Elsevier: Cambridge,2019; pp 153-172.
-
- 15. Chivukula, P.; Dušek, K.; Wang, D.; Dušková-Smrčková, M.; Kopečková, P.; Kopeček, J. Synthesis and Characterization of Novel Aromatic Azo Bond-containing pH-sensitive and Hydro- lytically Cleavable IPN Hydrogels. Biomaterials 2006, 27, 1140-1151.
-
- 16. Sperling, L. H. Interpenetrating Polymer Networks. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons: New York, 2004; pp 272-311.
- 17. Goujon, L. J.; Khaldi, A.; Maziz, A.; Plesse, C.; Nguyen, G. T.; Aubert, P. H.; Vidal, F.; Chevrot, C.; Teyssié, D. Flexible Solid Polymer Electrolytes Based on Nitrile Butadiene Rubber/poly (ethylene oxide) Interpenetrating Polymer Networks Containing Either LiTFSI or EMITFSI. Macromolecules 2011, 44, 9683-9691.
-
- 18. Kaczmarek, H.; Vuković-Kwiatkowska, I. Preparation and characterization of Interpenetrating Networks Based on Poly- acrylates and Poly(lactic acid). Exp. Polym. Lett. 2018, 6, 78-94.
-
- 19. Myung, D.; Waters, D.; Wiseman, M.; Duhamel, P. E.; Noolandi, J.; Ta, C. N.; Frank, C. W. Progress in the Development of Interpenetrating Polymer Network Hydrogels. Polym. Adv. Technol. 2008, 19, 647-657.
-
- 20. Wang, J. J.; Liu, F. Enhanced Adsorption of Heavy Metal Ions Onto Simultaneous Interpenetrating Polymer Network Hydrogels Synthesized by UV Irradiation. Polym. Bulletin 2013, 70, 1415-1430.
-
- 21. Apopei, D. F.; Dragan, E. S. Semi-interpenetrating Polymer Networks based on Polyacrylamide and Starch or Modified Starch. J. Nanostruct. Polym. Nanocompos. 2013, 9, 16-20.
- 22. Dragan, E. S.; Apopei, D. F. Multiresponsive Macroporous Semi-IPN Composite Hydrogels Based on Native or Anionically Modified Potato Starch. Carbohydr. Polym. 2013, 92, 23-32.
-
- 23. Dragan, E. S.; Loghin, D. F. A. Enhanced Sorption of Methylene Blue From Aqueous Solutions by semi-IPN Composite Cryogels with Anionically Modified Potato Starch Entrapped in PAAm Matrix. Chem. Eng. J. 2013, 234, 211-222.
-
- 24. Dinu, M. V.; Perju, M. M.; Drăgan, E. S. Composite IPN Ionic Hydrogels Based on Polyacrylamide and Dextran Sulfate. React. Funct. Polym. 2011, 71, 881-890.
-
- 25. Dinu, M. V.; Schwarz, S.; Dinu, I. A.; Drăgan, E. S. Comparative Rheological Study of Ionic semi-IPN Composite Hydrogels Based on Polyacrylamide and Dextran Sulphate and of Polyacrylamide Hydrogels. Colloid Polym. Sci. 2012, 290, 1647-1657.
-
- 26. Chirila, T. V.; George, K. A.; Abdul Ghafor, W. A.; Pas, S. J.; Hill, A. J. Sequential Homo‐interpenetrating Polymer Networks of Poly(2‐hydroxyethyl methacrylate): Synthesis, Characterization, and Calcium Uptake. J. Appl. Polym. Sci. 2012, 126, E455-E466.
-
- 27. Lee, Y.; Kim, D. N.; Choi, D.; Lee, W.; Park, J.; Koh, W. G. Preparation of Interpenetrating Polymer Network Composed of Poly(ethylene glycol) and Poly(acrylamide) Hydrogels as a Support of Enzyme Immobilization. Polym. Adv. Technol. 2008, 19, 852-858.
-
- 28. Liu, Y. Y.; Lü, J.; Shao, Y. H. Preparation and Characterization of Poly(N‐isopropylacrylamide)‐modified Poly(2‐hydroxyethyl acrylate) Hydrogels by Interpenetrating Polymer Networks for Sustained Drug Release. Macromol. Biosci. 2006, 6, 452-458.
-
- 29. Lin, M. S.; Liu, C. C.; Lee, C. T. Toughened Interpenetrating Polymer Network Materials Based on Unsaturated Polyester and Epoxy. J. Appl. Polym. Sci. 1999, 72, 585-592.
-
- 30. Guhanathan, S.; Hariharan, R.; Sarojadevi, M. Studies on Castor Oil-based Polyurethane/polyacrylonitrile Interpenetrating Polymer Network for Toughening of Unsaturated Polyester Resin. J. Appl. Polym. Sci. 2004, 92, 817-829.
-
- 31. Chung, J. W.; Park, J. H.; Choi, H. M.; Oh, K. W. Synthesis and Characterization of a Dyeable Bio-based Polyurethane/branched Poly(ethylene imine) Interpenetrating Polymer Network with Enhanced Wet Fastness. Text. Res. J. 2019, 89, 335-346.
-
- 32. Fan, H.; Gong, J. P. Fabrication of Bioinspired Hydrogels: Challenges and Opportunities. Macromolecules 2020, 53, 2769-2782.
-
- 33. Haddrick, M.; Simpson, P. B. Organ-on-a-chip Technology: Turning its Potential for Clinical Benefit into Reality. Drug Discovery Today 2019, 24, 1217-1223.
-
- 34. Hoffman, T.; Khademhosseini, A.; Langer, R. Chasing the Paradigm: Clinical Translation of 25 Years of Tissue Engineering. Tissue Eng. Part A 2019, 25, 679-687.
-
- 35. Che, Y.; Li, D.; Liu, Y.; Yue, Z.; Zhao, J.; Ma, Q.; Zhang, Q.; Tan, Y.; Yue, Q.; Meng, F. Design and Fabrication of a Triple-responsive Chitosan-based Hydrogel with Excellent Mechanical Properties for Controlled Drug Delivery. J. Polym. Res. 2018, 25, 169.
-
- 36. Zoratto, N.; Matricardi, P. Semi-IPNs and IPN-based Hydrogels. In Polymeric Gels; Pal, K., Banerjee, I., Eds.; Woodhead Publishing: Cambridge,2018; pp 91-124.
-
- 37. Park, S. H.; Shin, H. S.; Park, S. N. A Novel pH-responsive Hydrogel Based on Carboxymethyl Cellulose/2-hydroxyethyl Acrylate for Transdermal Delivery of Naringenin. Carbohydr. Polym. 2018, 200, 341-352.
-
- 38. Mahou, R.; Vlahos, A. E.; Shulman, A.; Sefton, M. V. Interpenet- rating Alginate-collagen Polymer Network Microspheres for Modular Tissue Engineering. ACS Biomater. Sci. Eng. 2017, 4, 3704-3712.
-
- 39. Soni, S. R.; Bhunia, B. K.; Kumari, N.; Dan, S.; Mukherjee, S.; Mandal, B. B.; Ghosh, A. Therapeutically Effective Controlled Release Formulation of Pirfenidone from Nontoxic Biocompatible Carboxymethyl Pullulan-Poly(Vinyl Alcohol) Interpenetrating Polymer Networks. ACS Omega 2018, 3, 11993-12009.
-
- 40. Ji, W. G.; Hu, J. M.; Liu, L.; Zhang, J. Q.; Cao, C. N. Water Uptake of Epoxy Coatings Modified with γ-APS Silane Monomer. Prog. Org. Coat. 2006, 57, 439-443.
-
- 41. Montemor, M. F.; Ferreira, M. G. S. Electrochemical Study of Modified Bis-[Triethoxysilylpropyl] Tetrasulfide Silane Films Applied on the AZ31 Mg Alloy. Electrochim. Acta. 2007, 52, 7486-7495.
-
- 42. Qian, M.; Soutar, A. M.; Tan, X. H.; Zeng, X. T.; Wijesinghe, S. L. Two-part Epoxy-siloxane Hybrid Corrosion Protection Coatings for Carbon Steel. Thin Solid Films 2009, 517, 5237-5242.
-
- 43. Abe, Y.; Gunji, T. Oligo- and Polysiloxanes. Prog. Polym. Sci. 2004, 29, 149-182.
-
- 44. Chauhan, B. P.; Balagam, B. Silyl Functionalization of Polyolefins. Macromolecules 2006, 39, 2010-2012.
-
- 45. Chen, Y.; Kim, H. Poly(vinylidene fluoride) Grafted with 3-trimethoxysilylpropyl Methacrylate for Silyl Functional Membranes. React. Funct. Polym. 2008, 68, 1499-1506.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2021; 45(6): 857-864
Published online Nov 25, 2021
- 10.7317/pk.2021.45.6.857
- Received on Apr 21, 2021
- Revised on Jun 17, 2021
- Accepted on Jun 21, 2021
Services
Shared
Correspondence to
- Firas Jameel Jabbar
-
Department of Pharmacy, Southern Technical University, Basrah, Iraq
- E-mail: firas.alassadi@stu.edu.iq
- ORCID:
0000-0002-6928-9450
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr