






- Conjugated Polyelectrolyte-Based Fluorescent Surfactants: Synthesis and Emulsion Formation
Byungjin Koo†

Department of Polymer Science and Engineering, Dankook University, 152, Jukjeon-ro, Suji-gu, Yongin-si, Gyeonggi-do 16890, Korea
- 공액 고분자 전해질 기반 계면활성제: 합성 및 에멀젼 형성
구병진†
단국대학교 고분자공학과
Reproduction, stored in a retrieval system, or transmitted in any form of any part of this publication is permitted only by written permission from the Polymer Society of Korea.
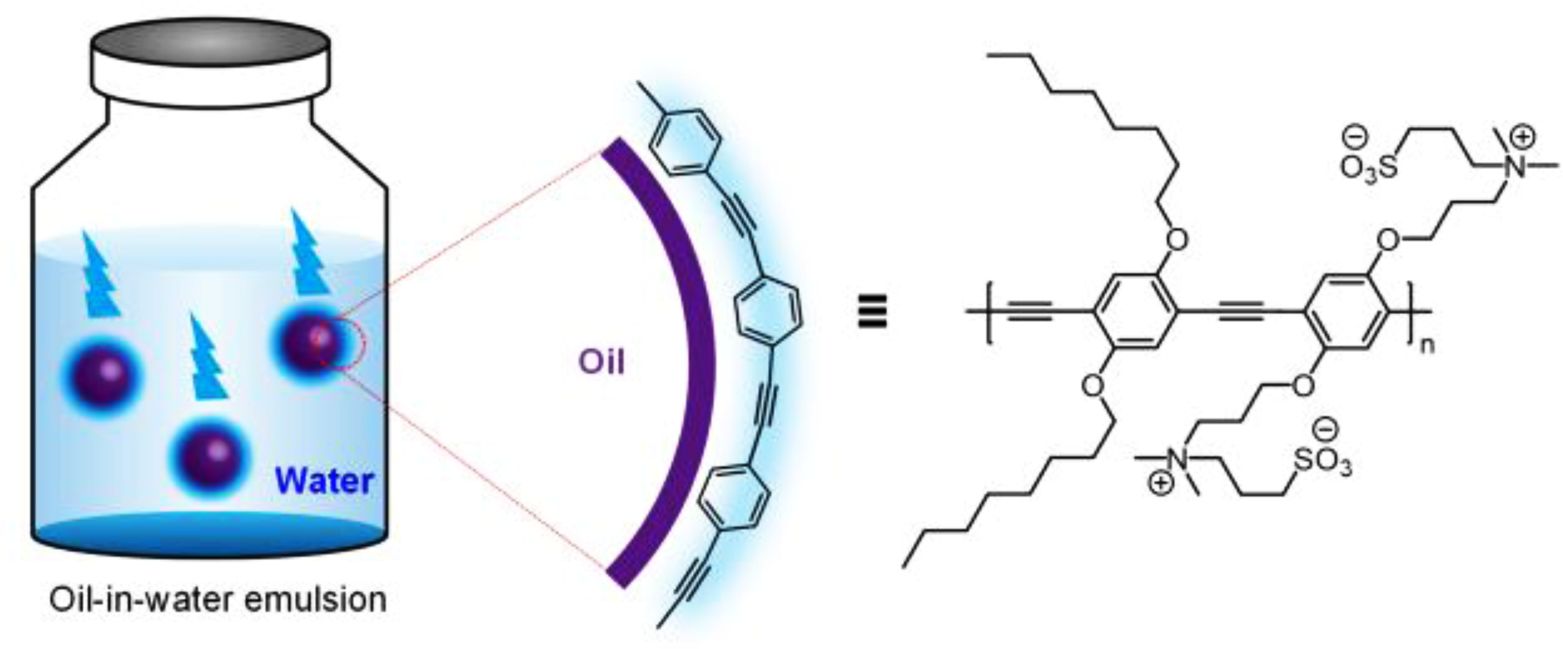
Visualization of the amphiphilic interface through fluorescence is of importance to understand the interfacial phenomena in a non-invasive way. However, there is a scarcity of surfactant conjugated polymers with strong fluorescence owing to the difficulty of synthetic procedures employing a hydrophobic polymer backbone with strongly hydrophilic side-chains. Herein, we report novel fluorescent polymeric surfactants based on conjugated polyelectrolytes (CPEs) containing zwitterionic and alkyl side-chains. A fluorescent poly(phenylene ethynylene) (PPE) backbone with amine and alkyl groups was synthesized by Sonogashira-type polymerization. The amines on PPE were treated with 1,3-propanesultone, resulting in the formation of zwitterionic groups. Stable oil-in-water emulsion was successfully formed with co-surfactants. The conformations of polymers in different physical states were compared through photophysical analysis. It was found that the PPE backbone was highly planarized at the oil-water interface with extended π-π interactions, providing potential applications in the non-invasive visualization of amphiphilic interfaces found in colloids and biology.
양극성 계면을 형광을 통해 시각화하는 것은 계면에서 발생하는 여러 과학적 현상을 이해하는 데에 중요하다. 하지만 양극성 및 형광 특성을 동시에 보여주는 고분자의 종류는 매우 적으며, 그 주된 이유는 형광을 보이는 소수성 고분자 주사슬과 강한 친수성을 가지는 곁사슬을 동시에 합성하는 것이 까다롭기 때문이다. 공액 고분자 전해질 구조를 기반으로, 폴리페닐렌 에타이닐렌을 주사슬로 하는 새로운 양극성 형광 고분자를 합성하였다. 각각 아민과 알킬그룹을 가지는 두 단량체를 소노가시라 중합법을 통해 고분자를 합성하고, 1,3-프로판설톤과의 반응을 통해 아민그룹을 양쪽성이온(zwitterion)으로 만들어 강한 극성을 가지도록 하였다. 공계면활성제를 함께 이용하여 안정적인 오일-물 에멀젼을 형성하였다. 고분자의 형태를 광물리학을 이용해 분석한 결과, 용액, 침전, 박막과 비교하여 에멀젼에서는 고분자의 형태가 크게 평면화되고 따라서 π-π 상호작용이 커짐을 발견하였다. 이러한 결과는 향후 다양한 콜로이드 및 생물학에서 나타나는 양극성 계면의 시각화에 응용될 수 있을 것으로 기대된다.
A conjugated polyelectrolyte-based fluorescent surfactant was synthesized through Sonogashira-type polymerization and subsequent zwitterionization. This polymer with a co-surfactant could stabilize oil droplets in water. The polymer was localized at the interface, resulting in the planarization of the polymer backbone with the long-range π-π interactions. The interfacial localization of the fluorescent polymer could provide a new way to visualize amphiphilic interfaces in a non-invasive way.
Keywords: fluorescent polymer, conjugated polymer, conjugated polyelectrolyte, surfactant, emulsion.
I thank Prof. Timothy Swager at MIT for helpful discussion. The present research was supported by the research fund of Dankook University in 2020.
NMR spectra of monomers and polymers are available via the Internet at http://journal.polymer-korea.or.kr.
PK_2021_045_04_641_Supporting_Information_template.pdf (291 kb)
Supplementary Information
Surfactants have been extensively utilized in our daily lives and academic research such as in food, cosmetics, pharmaceuticals, and drug delivery.1,2 Fluorescent surfactants provide further benefits since they are able to visualize the amphiphilic interfaces in a non-invasive way, with potential applicability in colloidal systems and bioimaging. Only a few examples of fluorescent surfactants exist so far, and most of them are based on small molecule building blocks.3-7 Polymer-based fluorescent surfactants have also been developed over the past decades. They have advantages over low-molecular-weight counterparts in terms of high extinction coefficients and quantum yields that result in bright fluorescence, as well as structural complexities and nanostructures which are originated from large macromolecules.8 For example, the Swager group reported several surfactant-type poly(phenylene ethynylene)s (PPEs) that can be localized at the air-water interface or can stabilize oil droplets in water.9-11 These PPEs have different backbone conformation with different optical properties between bulk (3D) and interface (2D) owing to the varied degree of spatial confinement, which differentiates them from the corresponding small-molecule based fluorescent surfactants.
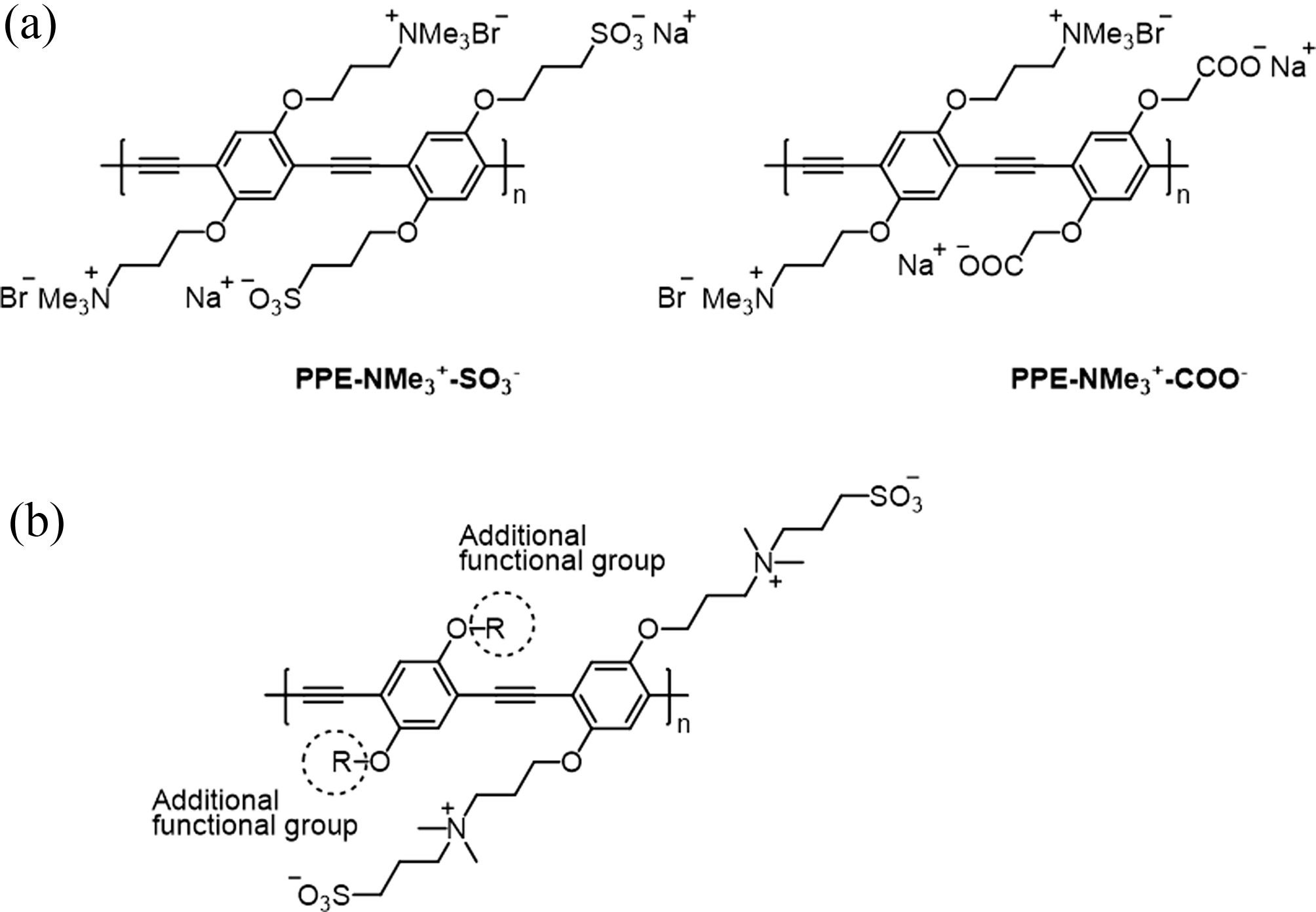
Fluorescent polymeric surfactants require a luminescent main-chain with both ionic and alkyl side-chains. These surfactants are derivatives of conjugated polyelectrolytes (CPEs) which possess a conjugated backbone and ionic side-chains. In particular, CPEs are versatile materials incorporated in various optoelectronic devices, wherein interfacial engineering with CPEs confers advantages such as tuning the work function and interfacial energy barriers.12-15 With the broad utility of CPEs in mind, we were interested in the synthesis of amphiphilic, fluorescent CPE-based materials by attaching additional alkyl chains to them. Previously, the Schanze group and others reported various types of CPEs,16 and we chose to focus on the polyampholytes17 that have both cationic and anionic functional groups in different repeat units, as shown in Figure 1 (PPE-NMe3+-SO3- and PPE-NMe3+-COO-). This polymer indicated pH-responsiveness due to carboxylates, which, on the other hand, could imply that the polymers may not be stable as surfactants in various pH.
In order to synthesize stable surfactant-type CPEs, zwitterionic side-chains may be more desired. Zwitterions have advantages in that they have fixed, permanent charges that are robust in different conditions such as various pH and temperature. In addition, zwitterions are known to have high water solubility and thus help hydrophobic moieties become compatible with water. Therefore, they have been employed in a variety of applications from nanotechnology to industry.18-21 To produce this zwitterion, we designed the target polymer as shown in Figure 1(b). In contrast to the previous polyampholyte-type polymers, the zwitterionic groups can make additional space for further derivatization (dashed circles in Figure 1), wherein alkyl chains can be introduced in this work, and various functions can also be appended for tailored applications.
|
Figure 1 Overview of this work: (a) Previously, PPE-NMe3 + -SO3 - and PPE-NME3 + -COO- were reported as polyampholytes; (b) In this work, one of the ionic groups can be moved to form zwitterions, providing an empty space for further derivatization with alkyl groups (dashed circles). |
General. Chemicals were purchased from Aldrich (USA), Alfa Aesar (USA), and TCI (Japan) without further purification unless noted otherwise. Nanopure water (18.2 MΩ/cm) for preparing emulsions was obtained with a Fisher Scientific barnstead nanopure system (USA). All reactions were carried out under argon with standard Schlenk techniques. Monomer 4 from 1,4-dimethoxybenzene was synthesized by following the literature procedures or slight modifications thereof.22,23
All 1H NMR and 13C NMR spectra were reported in ppm on a Bruker Avance-400 (USA). 1H NMR was referenced to a chloroform peak (δ = 7.26 ppm) or a dimethyl sulfoxide (DMSO) peak (δ = 2.50 ppm). The multiplicity was reported as follows: s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, p = pentet, m = multiplet or unresolved, and br = broad. Coupling constants J were reported in Hz. 13C NMR was referenced to a chloroform peak (δ = 77.16 ppm). High-resolution mass spectra (HRMS) were obtained using electrospray ionization (ESI) or direct analysis in real time (DART).
THF gel permeation chromatography (GPC) was performed with a concentration of 0.5 mg/mL on an Agilent 1260 Infinity system equipped with three PLgel columns (103, 104, 105 Å) in series (USA), calibrated with monodisperse polystyrene standards.
UV-vis spectra were recorded on Agilent Cary 4000 spectrometer (USA) at room temperature. Fluorescence measurements were performed at room temperature with a Horiba Jobin Yvon SPEX Fluorolog-τ3 fluorimeter (model FL-321, 450 W Xenon lamp, Japan) using right-angle conformation for solution and front-face conformation for thin films. For sample preparations of photophysics measurements, the solutions were prepared in appropriate solvents (chloroform or DMSO, depending on the solubility) with 10-5-10-6 M. The solid state films were prepared on glass by spin-coating the chloroform or DMSO solution with the polymer (1-5 mg/mL), with 1000-3000 rpm for 60 s.
Dynamic light scattering (DLS) data were obtained on a NanoBrook Omni particle size analyzer (Brookhaven Instruments, USA) and analysed with Particle Solutions software (version 2.6).
Monomer Synthesis and Polymerization. 1,4-bis(3-bromopropoxy)-2,5-diiodobenzene (2): To the flame-dried Schlenk flask, 1,4-dihydroxy-2,5-diiodobenzene (1) (1.5 g, 4.14 mmol, 1 equiv) and dibromopropane (10.04 g, 49.74 mmol, 12 equiv) were added and dissolved in dimethyl formamide (DMF) (40 mL), followed by the addition of potassium carbonate (1.72 g, 12.43 mmol, 3 equiv). The mixture was stirred at 60 oC for 23 h. The solvent was evaporated in vacuo. Water and diethyl ether were added and the aqueous layer was extracted, and the combined organic layer was washed with water and brine. The organic phase was dried over MgSO4, filtered, and concentrated. The crude material was purified by column chromatography (hexane:DCM = 4:1), producing a white solid. 45% yield (1.13 g). 1H NMR (400 MHz, CDCl3) δ 7.21 (s, 2H), 4.09 (t, J = 5.6 Hz, 4H), 3.69 (t, J = 6.4 Hz, 4H), 2.41-2.27 (m, 4H). HRMS calculated for C12H14Br2I2O2 ([M]+) 603.7429; found, 603.7420.
3,3'-((2,5-diiodo-1,4-phenylene)bis(oxy))bis(N,N-dimethylpropan-1-amine) (3): To the flame-dried Schlenk flask, 1,4-bis(3-bromopropoxy)-2,5-diiodobenzene (2) (980 mg, 1.62 mmol, 1 equiv), potassium carbonate (897 mg, 6.5 mmol, 4 equiv), and potassium iodide (11 mg, 0.065 mmol, 0.04 equiv) were added. The mixture was dissolved in acetonitrile (50 mL), followed by the addition of dimethylamine (1.95 mL of 2 M solution in THF, 3.9 mmol, 2.4 equiv). The mixture was stirred at 70 oC for 24 h. After cooling down to room temperature, the solvent was evaporated in vacuo. Water and ethyl acetate were added, and the aqueous layer was extracted. The combined organic layer was washed with water and brine. The organic phase was dried over MgSO4, filtered, and concentrated. The crude material was purified by column chromatography (DCM:MeOH = 5:1 with 3% triethylamine added), after which the fractions were filtered with a 0.45 μm syringe filter. 59% yield (511 mg). 1H NMR (400 MHz, CDCl3) δ 7.20 (s, 2H), 3.99 (t, J = 6.2 Hz, 4H), 2.52 (t, J = 7.2 Hz, 4H), 2.28 (s, 12H), 2.03-1.92 (m, 4H). HRMS calculated for C16H27I2N2O2 ([M+H]+) 533.0162; found, 533.0152.
P1: To the flame-dried Schlenk flask, a dialkyne monomer 4 (115 mg, 0.3 mmol, 1 equiv), a dihalide monomer 3 (168 mg, 0.315 mmol, 1.05 equiv), Pd(PPh3)4 (17 mg, 0.015 mmol, 5 mol%), and CuI (5.7 mg, 0.03 mmol, 10 mol%) were added. The flask containing reagents was evacuated and back-filled with argon for 5 times. The dry morpholine (12 mL) which was degassed with argon for 30 min was added. The reaction mixture was stirred for 30 min at room temperature to completely dissolve all the materials, followed by stirring at 65 oC for 14 h and then at 75 oC for 70 h. After cooling down to room temperature, the crude mixture was concentrated in vacuo, diluted with DCM, and filtered through a short plug (1-2 cm) of silica gel. After being concentrated in vacuo, the crude mixture was precipitated in DCM/methanol and centrifuged 14000 rpm for 10 min. The precipitates were filtered and washed with methanol, producing a solid product (143 mg, 72% yield). 1H NMR (400 MHz, CDCl3) δ 7.05 (s, 2H), 7.02 (s, 2H), 4.10 (t, J = 6.0 Hz, 4H), 4.04 (t, J = 6.3 Hz, 4H), 2.56-2.47 (m, 4H), 2.25 (s, 12H), 2.06-1.97 (m, 4H), 1.89-1.80 (m, 4H), 1.63 (br, 4H), 1.27 (br, 16H), 0.89 – 0.81 (m, 6H). Proton numbers that are only in the repeat unit are indicated. GPC (THF): Mn = 1.9 kDa, Đ = 2.87, Mp = 3.9 kDa. Degree of polymerization = 3 (based on Mn) or 6 (based on Mp).
P2: To the flame-dried Schlenk flask, P1 (50 mg, 0.076 mmol based on repeat unit) was dissolved in THF (1 mL), followed by the addition of 1,3-propanesultone (22 mg, 0.016 mL, 0.18 mmol). The mixture was stirred at 40 oC for 14 h. After cooling down to room temperature, ethyl acetate was added to form precipitates. After centrifugation at 14000 rpm for 10 min, the precipitates were filtered, producing an orange solid (61 mg, 89% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.23 (s, 2H), 7.20 (s, 2H), 4.16 (br, 4H), 4.07 (br, 4H), 3.47 (br, 8H), 3.03 (br, 12H), 2.41 (br, 4H), 2.20 (br, 4H), 1.96 (br, 4H), 1.73 (br, 4H), 1.46 (br, 4H), 1.31-1.20 (br, 16H), 0.81 (br, 6H). Proton numbers that are only in the repeat unit are indicated.
Polymer Aggregation and Emulsion. For aggregation, P2 was first dissolved in DMSO. Then DMSO (0.1 mL) was injected into deionized water (2.9 mL), and absorption and emission spectra were taken. For emulsion, deionized water (4.5 mL, containing 5 mM SDS and 10 mM NaCl) and hexadecane (0.5 mL, containing P2) were mixed and vortexed for 1 min, after which fluorescent emulsion was formed. DLS and emission spectra at each time point (6, 18, 30, and 42 h) were taken. UV-vis spectra could not be taken due to the cloudiness of the emulsion.
The synthetic procedure to produce zwitterionic, fluorescent surfactant CPEs is outlined in Scheme 1. Similar to the previous work in Figure 1, we targeted PPE (specifically P2) as a fluorescent polymer scaffold because PPE has a high extinction coefficient and fluorescence quantum yield as a consequence of the direct optical bandgaps and rigid excited-state structures.24,25 The synthesis started with 1,4-dihydroxy-2,5-diiodobenzene (1) that underwent an alkylation reaction in the presence of potassium carbonate and excess 1,3-dibromopropane (Williamson ether synthesis). It is important to keep the 1,3-dibromopropane excess to prevent unwanted double substitution of 1,3-dibromopropane. Subsequent amination with N,N-dimethylamine produced a diamino compound (3) that would be able to undergo zwitterionization in the post-polymerization modification stage (vide infra).
A Sonogashira-type cross-coupling polymerization reaction was executed with dioctoxy-diethynylbenzene (4) and diamino-diiodobenzene (3) in the presence of Pd(PPh3)4 and CuI, resulting in the formation of P1. The dihalide monomer (3) was added with a 5% excess in order to keep the end-groups being aryl halides instead of alkynes since, if alkynes were added in excess, the residual copper and ambient oxygen present during the purification steps could trigger a Glaser-type coupling reaction between alkynyl polymer end-groups, increasing the polydispersity. The molecular weights (MW) of 1.9 kDa (number-average MW, Mn) and 3.9 kDa (peak MW, Mp) were obtained with the polydispersity of 2.89. These Mn and Mp correspond to the degree of polymerization of 3 (six of alkyne-Ph moieties) and 6 (twelve of alkyne-phenyl moieties), respectively. Although the molecular weight was not high, it is enough to perform a spectroscopic analysis because PPEs are known to have an effective conjugation length of 3 (six of alkyne-Ph moieties).26 This means that the current polymer can show photophysical properties (vide infra) that are equivalent to the PPEs with higher molecular weights.
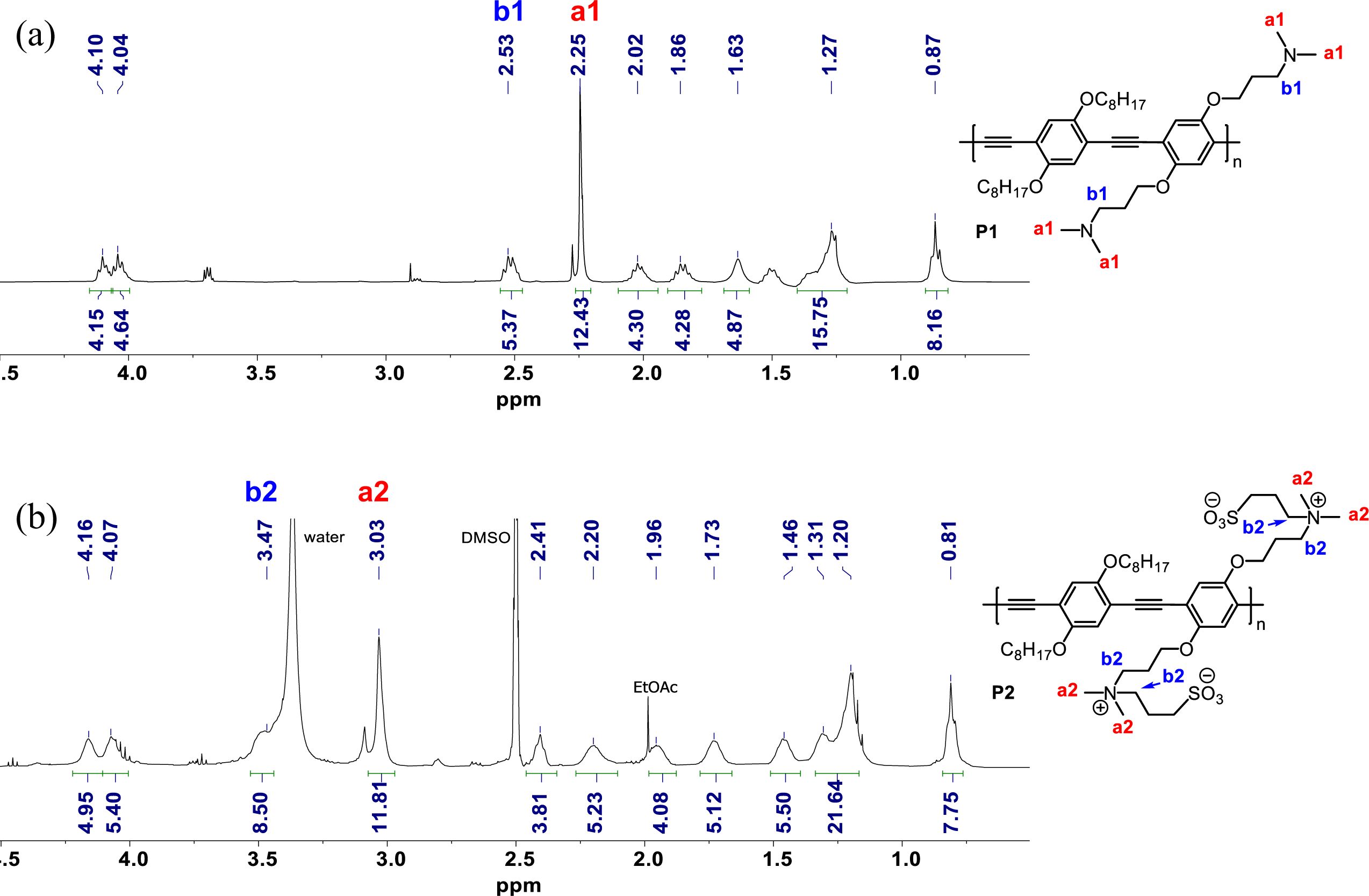
Quaternization of P1 proceeded in the presence of 1,3-propanesultone at 40 oC overnight, resulting in the formation of zwitterionic polymer P2. Zwitterionization was confirmed by nuclear magnetic resonance (NMR) spectroscopy, as shown in Figure 2 (full NMR spectra are displayed in Supporting Information). The characteristic signal transition upon zwitterionization can be examined by tracking the chemical shift of the two methyl groups (a1 in Figure 2) in the tertiary amine. Before quaternization, they appeared as one singlet at 2.25 ppm. After quaternization, as a consequence of the positive charges as well as the hyperconjugation effect, a downfield chemical shift was observed, resulting in the appearance of a singlet at 3.03 ppm (Figure 2(b)). The complete disappearance of the peak at 2.25 ppm indicates that the quaternization was quantitative. Another evidence of quaternization can be obtained from the chemical shift of b1 (2.53 ppm in Figure 2(a)) which moves to b2 (3.47 ppm in Figure 2(b)) upon quaternization due to the same reason as in a1.
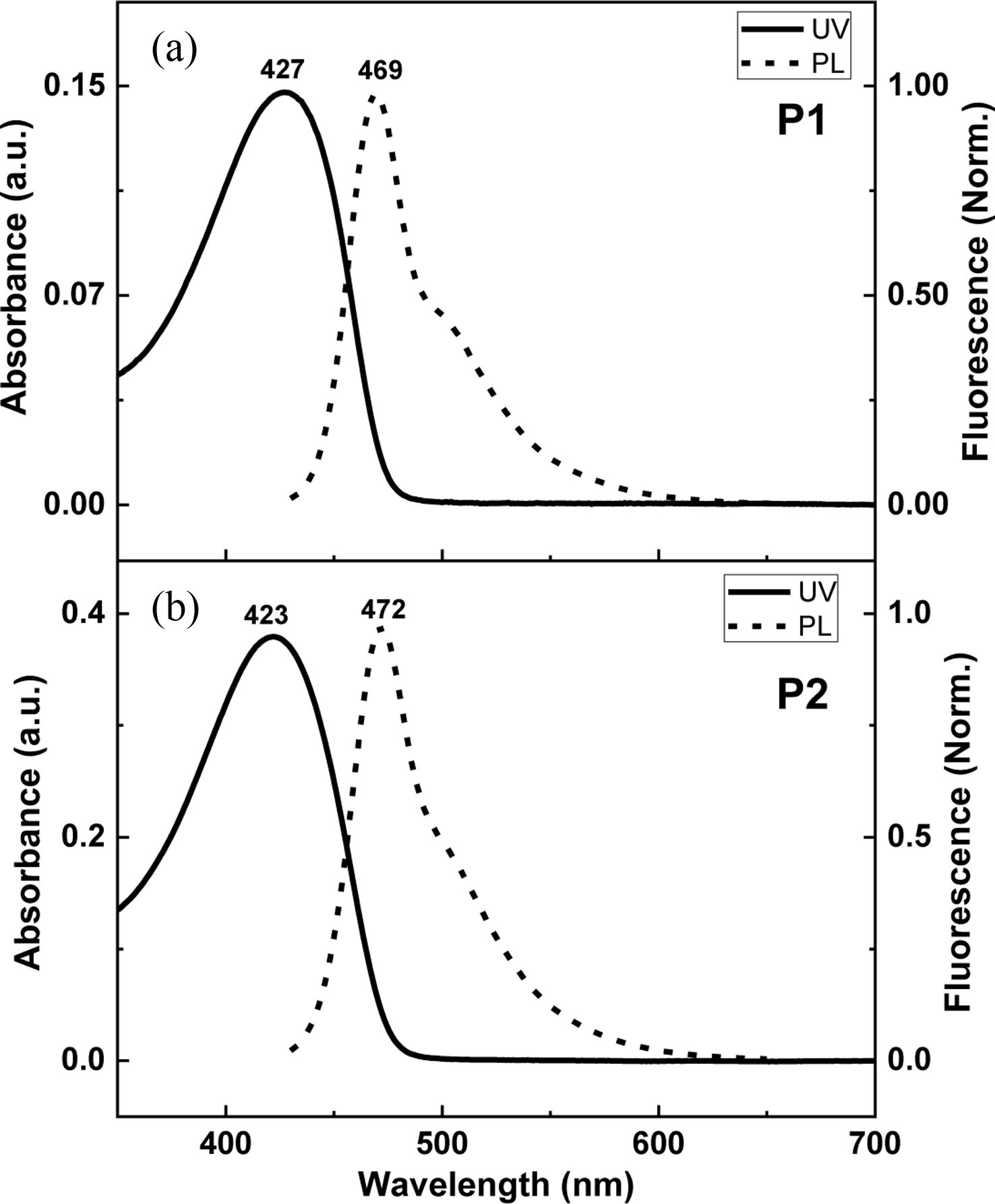
The absorption and emission spectra of P1 and P2 in the solution are displayed in Figure 3. P1 shows the absorption and emission maxima at 427 nm and 469 nm, respectively, with the Stokes shift of 42 nm. These values are consistent with the typical photophysical data from the PPE-type backbone,11 indicating that P1 has reached the effect conjugation length. P2 has the absorption and emission maxima at 423 nm and 472 nm, respectively. The Stokes shift of 49 nm was slightly higher than that of P1.
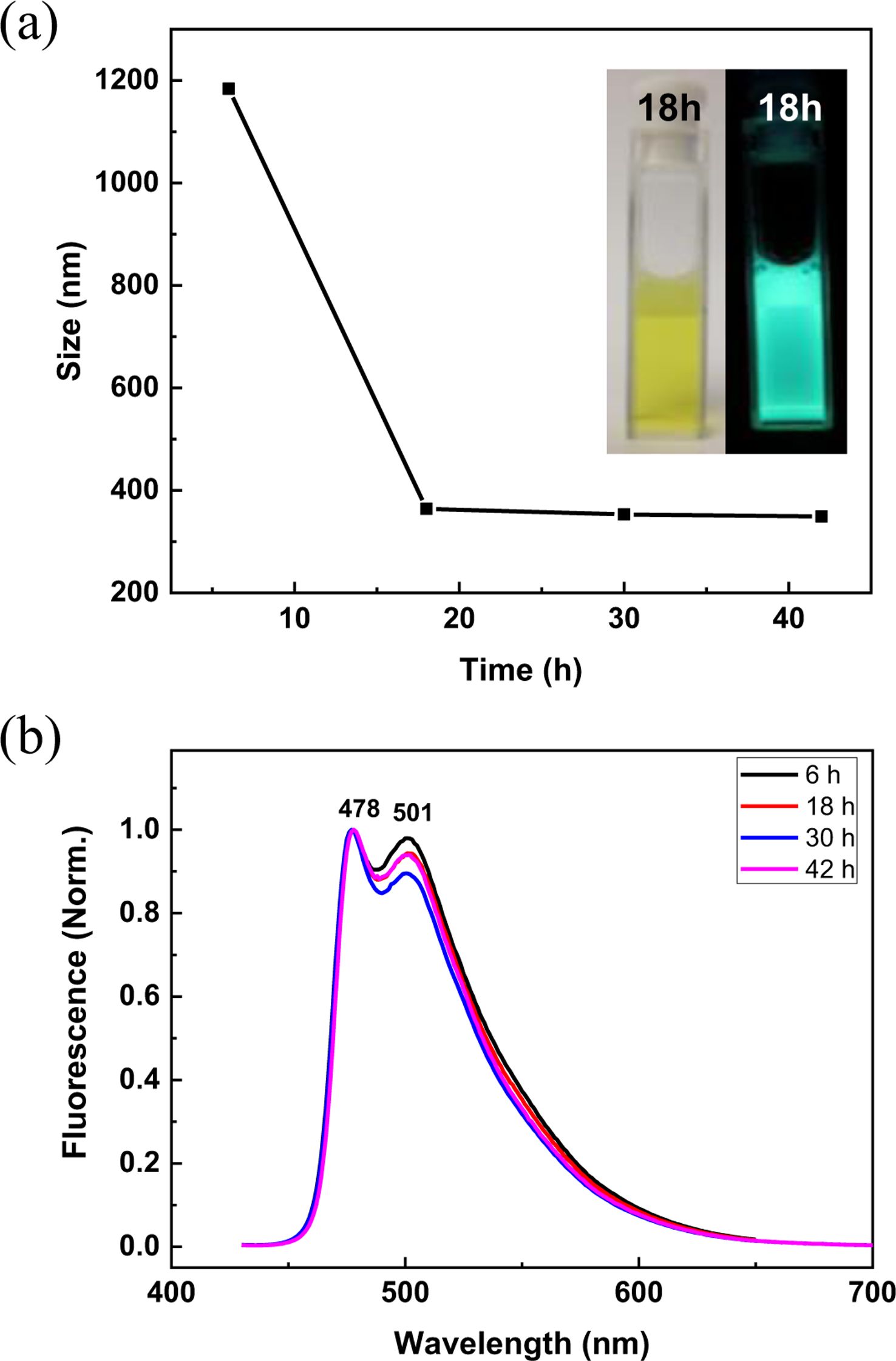
Next, the emulsion formation and related photophysics were studied. First, it was found that P2 by itself was not able to form a stable emulsion. The reason may be related to the diffusion kinetics of this polymeric surfactant, wherein generally the rate of adsorption of polymeric surfactants to the oil-water interface is believed to be slower than the collision between oil droplets, resulting in the coalescence of droplets. On the other hand, small molecular emulsifiers are believed to diffuse faster to form stable droplets.27 A mixed surfactant strategy with small molecules and polymers was implemented so that there would be enough time for polymeric surfactants to be adsorbed at the interface. Therefore, sodium dodecyl sulfate (SDS) was used as a co-surfactant because it is one of the most widely used ionic surfactants and is known to form stable oil-in-water emulsions.28 Emulsion was prepared by taking 4.5 mL of 5 mM SDS and 10 mM NaCl in water, to which 0.5 mL of hexadecane containing P2 was added. The mixture was vortexed for 1 min, and the size of emulsion as well as photoluminescence spectra were recorded at each time point (Figure 4). The sizes of droplets were each at around 1200 nm at 6 h and maintained around 350 nm after 18 h of incubation. The inset displays the picture of the emulsion sample at 18 h before (left) and after (right) the irradiation under a handheld UV lamp at 365 nm. Since the sizes of the droplets were less than the wavelength of visible light, they looked relatively transparent. Emission spectra of this emulsion sample at each time point are displayed in Figure 4(b). The most probable transition (0-0) was observed at 478 nm in all experimental times, which was 6 nm red-shifted from that of the solution, indicating that polymer backbones are planarized at the oil-water interface. In particular, the emission transition at 501 nm was also substantially increased compared to the solution, implying that the planarization and concomitant interpolymer interaction (π-π electronic communication) were highly significant.29-31 Slight fluctuation of this peak over time was observed, but the exact reason is currently unrevealed.
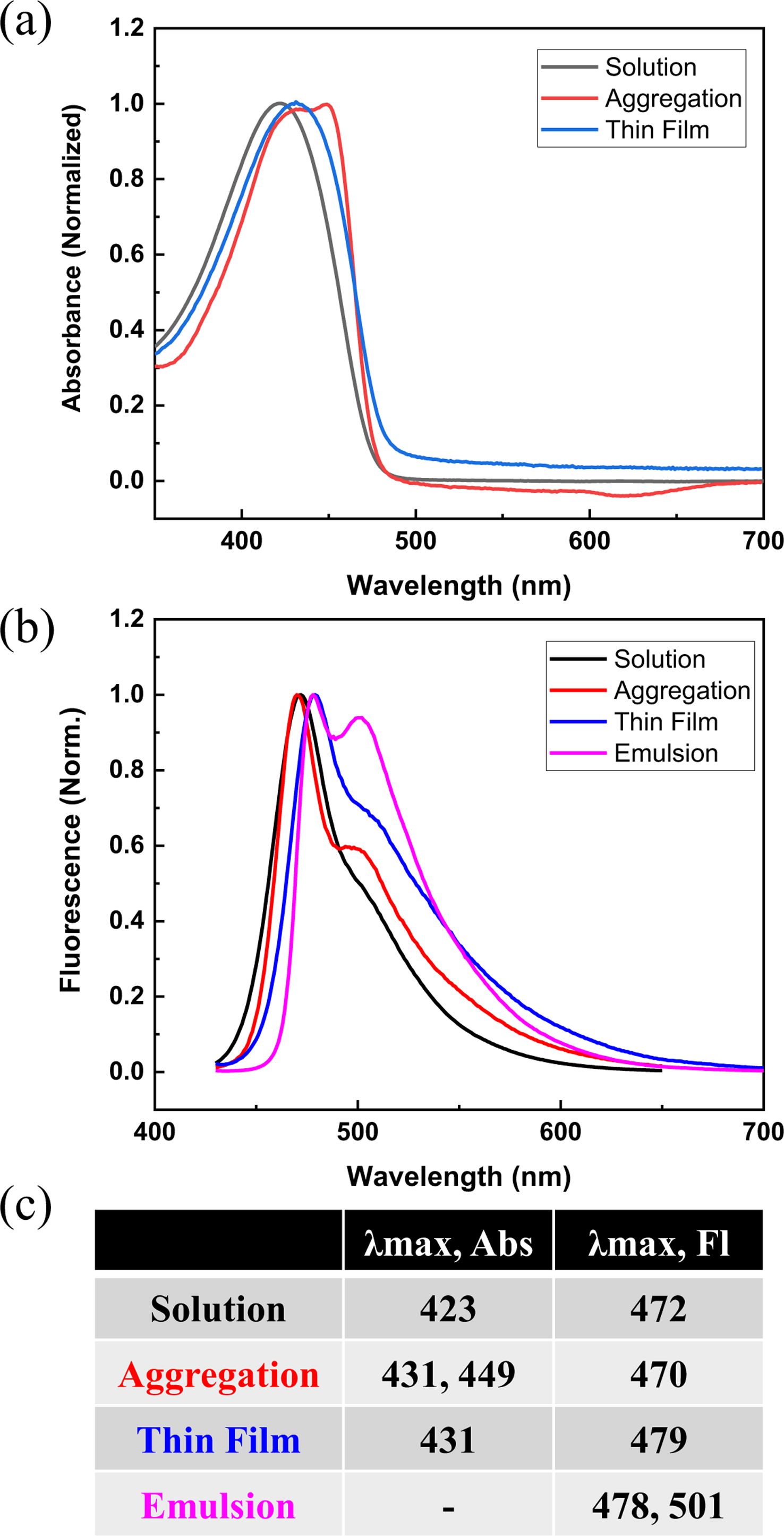
In order to further investigate the different physical states of P2, we prepared aggregation in solution and thin films and the corresponding absorption and emission spectra were measured (Figure 5). The aggregation was formed by taking 0.1 mL of P2 in DMSO (good solvent) and adding 2.9 mL of water (poor solvent). The absorption spectrum shows one maximum at 431 nm with another red-shifted maximum at 449 nm, which indicates the planarization of PPE backbone from H-type aggregates. The emission peak was observed at 470 nm, which is similar to the solution, although the quantum yield would be very low. Owing to the aggregation-induced fluorescence quenching, this 470 nm signal is presumably originated from the residual P2 dissolved in the solution (3.3% DMSO in water), not from the aggregates. For thin films, the absorption and emission maxima were recorded at 431 nm and 479 nm, respectively. These red-shifted maxima compared to the solution indicate that the planarization is induced in the solid state.
Regarding the comparison of the emulsion and thin films, the emission maxima were quite similar to each other, except the peak at 501 nm from the emulsion. This signal could suggest that P2 would adopt planar conformation as well as form strong π-π interactions because polymer chains have enough time to diffuse to the oil-water interface. On the other hand, P2 in thin films would be in a kinetically trapped state, wherein the planarization did occur but long-range interpolymer π-π interactions might not be formed. 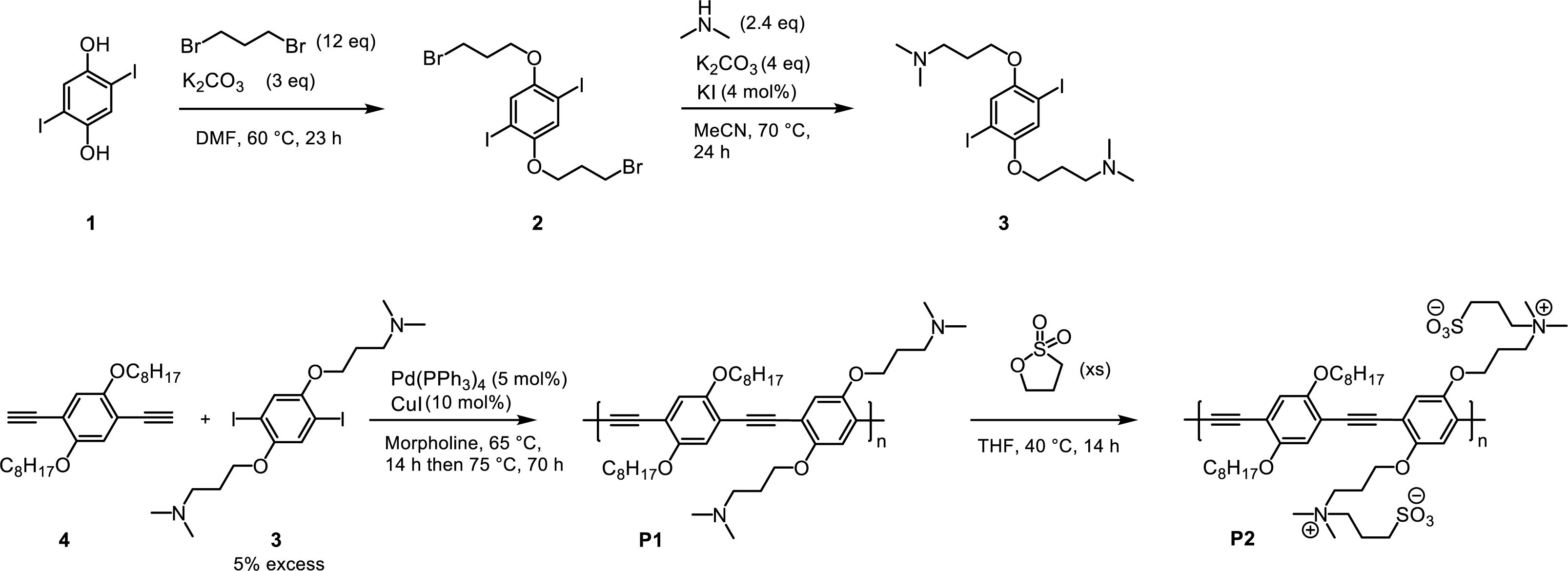
Scheme 1. Reaction steps to synthesize P2.
|
Figure 2 Partial 1H NMR spectra of (a) P1; (b) P2. |
|
Figure 3 Photophysical data of (a) P1; (b) P2 in solution. |
|
Figure 4 Characterization of the emulsion prepared from P2 with SDS as a co-surfactant: (a) the size of the emulsion over time with photographs of the emulsion at 18 h, before (left) and after (right) irradiation at 365 nm; (b) emission spectra at each time point. |
|
Figure 5 Photophysical properties of P2 in different states (solution, aggregation, thin film, and emulsion): (a) absorption spectra; (b) emission spectra; (c) tabulation of absorption and emission maxima. |
We herein report the synthesis of zwitterionic fluorescent CPEs and their application in oil-in-water emulsion formation. The polymer precursor P1 has both alkyl and amino groups, and subsequently the amino groups underwent zwitterionization with 1,3-propanesultone. The NMR analysis demonstrated the quantitative zwitterionization of P1. With SDS as a co-surfactant, stable emulsion with P2 was formed, wherein P2 was successfully localized at the oil-water interfaces. The localization and related photophysical properties from the emulsion were compared with solution, aggregation, and thin films of P2, indicating that the strong, long-range π-π interaction was present at the interface of emulsion. We believe that this work suggests a new class of CPE-based fluorescent surfactants for interfacial visualization and for future applications in which the understanding of interfacial phenomena is critical, such as in colloids and biology.
- 1. Myers, D. Surfactant Science and Technology; J. Wiley: Hoboken, USA, 2006.
-
- 2. Müllertz, A.; Ogbonna, A.; Ren, S.; Rades, T. New Perspectives on Lipid and Surfactant Based Drug Delivery Systems for Oral Delivery of Poorly Soluble Drugs. J. Pharm. Pharmacol. 2010, 62, 1622-1636.
-
- 3. Fallest, D. W.; Lichtenberger, A. M.; Fox, C. J.; Daniels, K. E. Fluorescent Visualization of a Spreading Surfactant. New J. Phys. 2010, 12, 073029.
-
- 4. Thijssen, J. H. J.; Schofield, A. B.; Clegg, P. S. How do (fluore- scent) Surfactants Affect Particle-stabilized Emulsions? Soft Matter 2011, 7, 7965-7968.
-
- 5. Yin, P.; Wu, P.; Xiao, Z.; Li, D.; Bitterlich, E.; Zhang, J.; Cheng, P.; Vezenov, D. V.; Liu, T.; Wei, Y. A Double-Tailed Fluorescent Surfactant with a Hexavanadate Cluster as the Head Group. Angew. Chem. Int. Ed. 2011, 50, 2521-2525.
-
- 6. Chen, C. V. H. H.; Liu, Y.; Stone, H. A.; Prud’homme, R. K. Visualization of Surfactant Dynamics to and Along Oil-water Interfaces Using Solvatochromic Fluorescent Surfactants. Langmuir 2018, 34, 10512-10522.
-
- 7. Wang, H.; Zhao, W.; Liu, X.; Wang, S.; Wang, Y. BODIPY-based Fluorescent Surfactant for Cell Membrane Imaging and Photo- dynamic Therapy. ACS Appl. Bio. Mater. 2020, 3, 593-601.
-
- 8. Raffa, P.; Wever, D. A. Z.; Picchioni, F.; Broekhuis, A. A. Poly- meric Surfactants: Synthesis, Properties, and Links to Applications. Chem. Rev. 2015, 115, 8504-8563.
-
- 9. Kim, J.; Swager, T. M. Control of Conformational and Interpolymer Effects in Conjugated Polymers. Nature 2001, 411, 1030-1034.
-
- 10. Koo, B.; Swager, T. M. Interfacial Pressure/area Sensing: Dual-fluorescence of Amphiphilic Conjugated Polymers at Water Interfaces. ACS Macro Lett. 2017, 6, 134-138.
-
- 11. Koo, B.; Swager, T. M. Distinct Interfacial Fluorescence in Oil-in-water Emulsions via Exciton Migration of Conjugated Polymers. Macromol. Rapid Commun. 2017, 38, 1700262.
-
- 12. Hoven, C. V.; Garcia, A.; Bazan, G. C.; Nguyen, T. Q. Recent Applications of Conjugated Polyelectrolytes in Optoelectronic Devices. Adv. Mater. 2008, 20, 3793-3810.
-
- 13. Huang, F.; Wu, H.; Cao, Y. Water/alcohol Soluble Conjugated Polymers as Highly Efficient Electron Transporting/injection Layer in Optoelectronic Devices. Chem. Soc. Rev. 2010, 39, 2500-2521.
-
- 14. Duarte, A.; Pu, K. Y.; Liu, B.; Bazan, G. C. Recent Advances in Conjugated Polyelectrolytes for Emerging Optoelectronic Appli- cations. Chem. Mater. 2011, 23, 501-515.
-
- 15. Cui, Q.; Bazan, G. C. Narrow Band Gap Conjugated Poly- electrolytes. Acc. Chem. Res. 2018, 51, 202-211.
-
- 16. Jiang, H.; Taranekar, P.; Reynolds, J. R.; Schanze, K. S. Conjugated Polyelectrolytes: Synthesis, Photophysics, and Applications. Angew. Chem. Int. Ed. 2009, 48, 4300-4316.
-
- 17. Ji, E.; Whitten, D. G.; Schanze, K. S. pH-Dependent Optical Properties of a Poly(Phenylene Ethynylene) Conjugated Poly- ampholyte. Langmuir 2011, 27, 1565-1568.
-
- 18. Lowe, A. B.; McCormick, C. L. Synthesis and Solution Properties of Zwitterionic Polymers. Chem. Rev. 2002, 102, 4177-4190.
-
- 19. Laschewsky, A. Structures and Synthesis of Zwitterionic Polymers. Polymers 2014, 6, 1544-1601.
-
- 20. Blackman, L. D.; Gunatillake, P. A.; Cass, P.; Locock, K. E. S. An Introduction to Zwitterionic Polymer Behavior and Appli- cations in Solution and at Surfaces. Chem. Soc. Rev. 2019, 48, 757-770.
-
- 21. Li, M.; Zhuang, B.; Yu, J. Functional Zwitterionic Polymers on Surface: Structures and Applications. Chem. Asian J. 2020, 15, 2060-2075.
-
- 22. Stalder, R.; Xie, D.; Zhou, R.; Xue, J.; Reynolds, J. R.; Schanze, K. S. Variable-gap Conjugated Oligomers Grafted to CdSe Nano- crystals. Chem. Mater. 2012, 24, 3143-3152.
-
- 23. Shirai, Y.; Zhao, Y.; Cheng, L.; Tour, J. M. Facile Synthesis of Multifullerene-OPE Hybrids via in situ Ethynylation. Org. Lett. 2004, 6, 2129-2132.
-
- 24. McQuade, D. T.; Pullen, A. E.; Swager, T. M. Conjugated Polymer-based Chemical Sensors. Chem. Rev. 2000, 100, 2537-2574.
-
- 25. Andrew, T. L.; Swager, T. M. Structure-Property Relationships for Exciton Transfer in Conjugated Polymers. J. Polym. Sci. B Polym. Phys. 2011, 49, 476-498.
-
- 26. Meier, H.; Stalmach, U.; Kolshorn, H. Effective Conjugation Length and UV/vis Spectra of Oligomers. Acta Polym. 1997, 48, 379-384.
-
- 27. McClements, D. J.; Jafari, S. M. Improving Emulsion Formation, Stability and Performance Using Mixed Emulsifiers: A review. Adv. Colloid Interface Sci. 2018, 251, 55-79.
-
- 28. Llamas, S.; Santini, E.; Liggieri, L.; Salerni, F.; Orsi, D.; Cristofolini, L.; Ravera, F. Adsorption of Sodium Dodecyl Sulfate at Water-Dodecane Interface in Relation to the Oil in Water Emulsion Properties. Langmuir 2018, 34, 5978-5989.
-
- 29. Miteva, T.; Palmer, L.; Kloppenburg, L.; Neher, D.; Bunz, U. H. F. Interplay of Thermochromicity and Liquid Crystalline Behavior in Poly(p-phenyleneethynylene)s: π-π Interactions or Planarization of the Conjugated Backbone? Macromolecules 2000, 33, 652-654.
-
- 30. Kim, Y.; Bouffard, J.; Kooi, S. E.; Swager, T. M. Highly Emissive Conjugated Polymer Excimers. J. Am. Chem. Soc. 2005, 127, 13726-13731.
-
- 31. Tekin, E.; Wijlaars, H.; Holder, E.; Egbe, D. A. M.; Schubert, U. S. Film Thickness Dependency of the Emission Colors of PPE-PPVs in Inkjet Printed Libraries. J. Mater. Chem. 2006, 16, 4294-4298.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2021; 45(4): 641-648
Published online Jul 25, 2021
- 10.7317/pk.2021.45.4.641
- Received on Apr 29, 2021
- Revised on Jun 15, 2021
- Accepted on Jun 21, 2021
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
- Supporting Information
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Byungjin Koo
-
Department of Polymer Science and Engineering, Dankook University, 152, Jukjeon-ro, Suji-gu, Yongin-si, Gyeonggi-do 16890, Korea
- E-mail: bkoo@dankook.ac.kr
- ORCID:
0000-0003-0681-8084
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr