






- Screening of Polymer Additives in Poloxamer 407 Hydrogel Formulations for Intravesical Instillation: Evaluation of Mechanical Properties, Gel-forming Capacity, and Drug Release
Hee Mang Yang# , Yong Hoon Won# , Ho Yub Yoon, Chang Hyun Kim, Yoon Tae Goo, In Ho Chang*, and Young Wook Choi†

College of Pharmacy, Chung-Ang University, 84 Heuksuk-ro, Dongjak-gu, Seoul 06974, Korea
*College of Medicine, Chung-Ang University, 84, Heuksuk-ro, Dongjak-gu, Seoul 06974, Korea- 방광 내 주입용 폴록사머 407 하이드로젤 제형의 고분자 첨가제 스크리닝: 기계적 성질, 젤 형성 능력 및 약물 방출 평가
양희망# · 원용훈# · 윤호엽 · 김창현 · 구윤태 · 장인호* · 최영욱†
중앙대학교 약학대학, *중앙대학교 의과대학
Poloxamer 407 (PLX) hydrogel
has been used as a drug delivery system for intravesical instillation, but it
has a limitation of insufficient gel strength. Here, to modulate the viscosity
and strength of hydrogel, various polymers were screened. Their effect on
viscosity decreased in the following order: high molecular-weight hyaluronic
acid (HHA; 33.524 Pa·s) > hydroxypropyl methylcellulose > chitosan >
low-molecular weight HA > sodium alginate > carbopol (24.332 Pa·s).
Polymer addition hardly altered the thermo-reversible property of hydrogels;
the gelation temperature was 21.0–25.3 oC and gelation time was
17.28–28.32 s. With gemcitabine as a water-soluble ingredient, gel erosion and
drug release were examined using an in vitro bladder simulation model.
After four repeated cycles of filling and emptying, the remaining fraction of
HHA-added hydrogel and PLX hydrogel was 74.6% and 57.8%, respectively.
Furthermore, drug release was diffusion- and erosion-controlled. Thus,
HHA-added hydrogel is a promising system for intravesical instillation.
폴록사머 407(PLX)
하이드로젤은 방광 내 주입을 위한 전달 시스템으로 사용되고 있지만, 젤의 강도가 충분하지
않다는 한계가 있다. 하이드로젤의 점도 및 강도를 조절하기 위해, 다양한
고분자 첨가제를 선별하였다. 점도의 변화는 고분자량 히알루론산(HHA;
33.524 Pa·s) > 히드록시프로필메틸셀룰로스
> 키토산 > 저분자량 HA > 알긴산 > 카보폴(24.332 Pa·s) 순서로 관찰되었다. 고분자 첨가는 PLX 하이드로젤의 열 가역적 특성을 변화시키지 않았으며, 젤화 온도(21.0-25.3 °C) 및 젤화 시간(17.28-28.32초)을 나타내었다. 수용성인 젬시타빈을 탑재한 채로 방광 시뮬레이션 모델을 통해 젤 침식 및 약물 방출을 조사하였다. 4회 반복시험 후 남아있는 양을 관찰하였을 때 HHA첨가 하이드로젤
및 PLX 하이드로젤이 각각 74.6% 및 57.8%로 나타났으며, 약물 방출은 확산 및 침식으로 제어되었다. 따라서 HHA 첨가 하이드로젤은 방광 내 주입을 위한 유망한 시스템으로
판단된다
Poloxamer (PLX) hydrogels with or without high molecular
weight hyaluronic acid (HHA) showed thermo-reversible property. Addition of HHA
decreased the release of drug, gemcitabine (GEM).
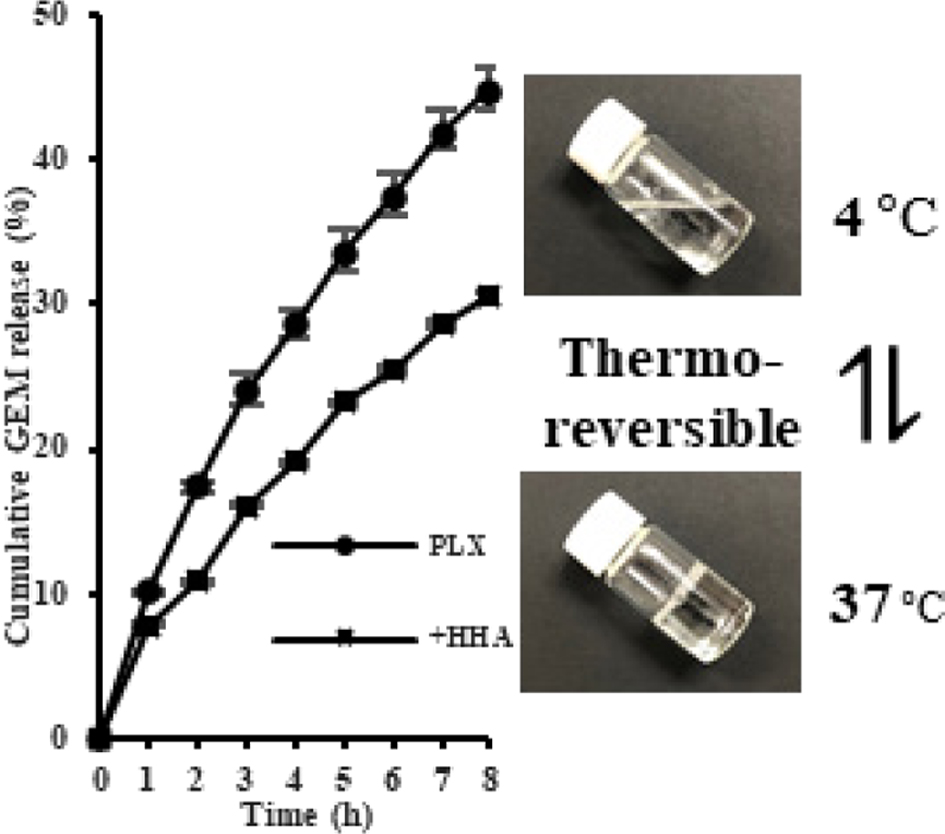
Keywords: poloxamer 407, hydrogel, gemcitabine, hyaluronic acid, intravesical instillation
This research was funded by the Ministry of Health and Welfare, Republic of Korea through the
Korea Health Industry Development Institute (KHIDI), grant
number HI17C0710. This research was also supported by the
Chung-Ang University Research Scholarship Grants in 2019.
Bladder cancer, one of the most common cancers worldwide,1 is
a heterogeneous disease, and 70% of patients have a superficial tumor with a
low risk of death and 30% have a life-threatening muscle-invasive tumor.2
The standard treatments for bladder cancer include radiotherapy, radical
cystectomy, immunotherapy, and chemotherapy. Various chemotherapeutic agents
such as paclitaxel, doxorubicin, and cisplatin are used as drugs of choice for
bladder cancer treatment. Recently, gemcitabine (GEM) has been commonly used in
combination or adjuvant chemotherapy for bladder cancer.3 However,
these agents generally fail to reach the therapeutic range when injected
intravenously. This limitation can be overcome by two strategies, namely, dose
increment and intravesical instillation. Because the former leads to systemic
adverse effects, the latter is a better option to treat bladder cancer.
However, intravesical instillation of a drug into the bladder using a catheter
has a limitation of dilution by urine and washout during urination. These
challenges can be overcome by employing in-situ gelling systems with
sol–gel transition property.
A hydrogel is a hydrophilic polymer network that contains a large amount
of water while maintaining its gel form.4,5 It possesses several
advantages such as diverse drug-incorporating capacity and in-situ
sol–gel transition after instillation in the body cavity. The typical mechanism
of sol–gel transition includes temperature- or pH-induced transition.6,7
Thermo-sensitive hydrogels exist in the hard-to-flow phase at the body
temperature of 37 oC or free-flowing phase at low temperature
(e.g., 4 oC).
Poloxamers, known as ABA-type triblock copolymers, consist of blocks of
ethylene oxide (EO) and propylene oxide (PO) units.8 One of the most
important characteristics of poloxamer is thermo-sensitiveness. Poloxamers have
a unique sol–gel transition temperature, at which they are in a flowable fluid
form, termed as sol. As the temperature increases, the poloxamer molecules
aggregate to form micelles, owing to the dehydration of the hydrophobic PO
blocks. The packing of these micelles, with the poly PO core and poly EO outer
shell, contribute to gelation, resulting in an un-flowable fluid, termed as
gel.9,10 Because of these characteristics, poloxamer hydrogel has
advantages as a delivery system for intravesical instillation; it is also less
toxic. Despite these merits, hydrogels composed of only poloxamers do not have
sufficient gel strength.
The addition of various excipients can modulate the viscosity and
strength of hydrogels.11 Neutral (carbopol, hydroxypropyl
methylcellulose, and hyaluronic acid), anionic (alginate, sodium
carboxymethylcellulose, and methacrylic acid copolymers), and cationic
(chitosan, chitin, and polyethylenimine) biocompatible polymers are commonly
used. The interactions between polymeric additives and poloxamers are known to
change the gel microstructure and rheological and mechanical properties. These
infrastructural and physicochemical changes could affect the gel-forming
capacity and the movement of drugs entrapped within the gel structure, thereby
changing the gel matrix erosion and drug release characteristics.12-16
In the present study, the strength of hydrogel was modulated by adding
selective polymers such as polysaccharide, cellulose derivatives and
carbohydrates. Mechanical properties of the modulated hydrogels were evaluated
in terms of viscosity, hardness, compressibility, adhesiveness, and
cohesiveness. Gel-forming capacity, in terms of gelation temperature and
gelation time, was compared among the hydrogels. Moreover, by adopting an in
vitro bladder simulation model, gel erosion and GEM release from the
hydrogels were investigated. We expect that these characteristics would be
favorable to develop a suitable drug delivery system for intravesical
instillation in bladder cancer treatment.
Materials. Poloxamer 407
(PLX) was supplied by BASF Laboratories (Wyandotte, MI, USA). Chitosan from
crab shells (CS; >400 mPa·s at 1% in acetic acid at 20 oC,
³75% deacetylated), hydroxypropyl methylcellulose (HPMC;
<26 kDa, 80–120 cps at 2% in water at 20 oC), sodium
alginate (ALG; 15–25 cps at 1% in water) and phosphate-buffered saline (PBS)
tablet were purchased from Sigma-Aldrich (St. Louis, MO, USA). Carbopol®
934NF (CP) was purchased from Lubrizol (Cleveland, OH, USA). Low-molecular
weight hyaluronic acid from cockscomb (LHA; 5–150 kDa) was purchased from TCI
(Toshima, Kita-ku, Tokyo, Japan). High-molecular weight hyaluronic acid (HHA; ³1000 kDa) was purchased from Thermo Fisher Scientific
(Ward Hill, MA, USA). Acetonitrile was purchased from J. T. Baker (Phillipsburg,
NJ, USA). GEM was provided by Shin Poong Pharm. Co., Ltd. (Gangnam, Seoul,
Korea). All other solvents used were of analytical grade.
Preparation
of PLX Hydrogel Samples. Thermo-reversible
PLX hydrogel was prepared using the cold technique.17 To prepare PLX
hydrogel without additives (reference sample), 20 w/v% PLX was added to
distilled water and stirred using a magnetic stirrer overnight in a
refrigerator (CLG-650; Jeio Tech, Daejeon, Korea) at 4 oC until
the PLX granules were completely dissolved. Polymer-added test samples were
prepared by adding the respective polymers (CS, ALG, HPMC, CP, LHA, and HHA) to
the PLX solution at 1 w/v% concentration, and then stirred overnight at
4 oC until a clear solution was obtained. GEM was loaded into
different hydrogels (1.5 mg/mL) and completely dissolved by stirring
overnight under the same conditions.
Visual
Observation of the Thermo-reversible Property. Five milliliters
of different hydrogels was loaded into transparent vials and stored at 4 oC
(in a refrigerator) and 37 oC (in a temperature-controlled
water bath, WB-11; Daihan Scientific, Wonju, Korea). The vials were then
tilted, and the degree of flowability was visually observed to confirm the
thermo-reversible transition from sol to gel.
Evaluation
of Mechanical Properties. The textural
properties of different hydrogels were measured using the TA-XT Express (Stable
Micro System Ltd., Surrey, UK). While avoiding air entrapment and making the
upper surface smooth, 25 mL of samples was filled in 50 mL beakers. When
each sample changes to the gel state at 37 oC, a 20 mm
(diameter) probe was inserted into the sample. The test speed rate, distance
(depth of insertion), and trigger force were set to 0.5 mm/s, 5 mm, and
1 g, respectively.18 The mechanical parameters of hydrogels,
such as hardness, compressibility, adhesiveness, and cohesiveness, were
determined from the force versus time plot. The maximum force reflects the
hardness of hydrogel, and it is defined as the force required to deform the sample.
The area of downward movement of the probe shows the compressibility of
hydrogel, whereas the area of upward movement of the probe shows the
adhesiveness of hydrogel. The proportional force of the first downward movement
to the second downward movement of the probe can be defined as cohesiveness.
Separately, the viscosity of hydrogels was measured using the Advanced
Rheometric Expansion System (ARES; Rheometric Scientific Inc., London, UK). In
the gel state of the samples, 2 mL of hydrogel was loaded into 25 mm
parallel plates of the ARES. With a single steady rate of 8 h-1 and
1 mm gap, the viscosity of each sample was measured while maintaining
37 oC.19 The texture analysis and viscosity
measurement were performed in triplicates and the values were averaged.
Measurement
of Gelation Temperature and Gelation Time. The gelation
temperature and gelation time were measured using the test-tube inversion
method.20,21 An aliquot (1 mL) of the sample was instilled into
5 mL test tubes (11 mm diameter) at 4 oC, as the sol
phase. The prepared test tubes containing hydrogel were placed in a
temperature-controlled water bath and the temperature was gradually increased
at the rate of 1 oC/min from 4 to 37 oC. The
temperature at which the sample does not flow for 30 s when the test-tube is
inverted is the gelation temperature. Separately, 1 mL of the sample was
injected into a 5 mL test tube at 4 oC as the sol phase,
and immediately, the test tube was placed in a water bath of 37 oC;
then, the gelation time was checked every 5 s until the sample was not
flowable, as the gel phase. The measurements were performed in triplicate.
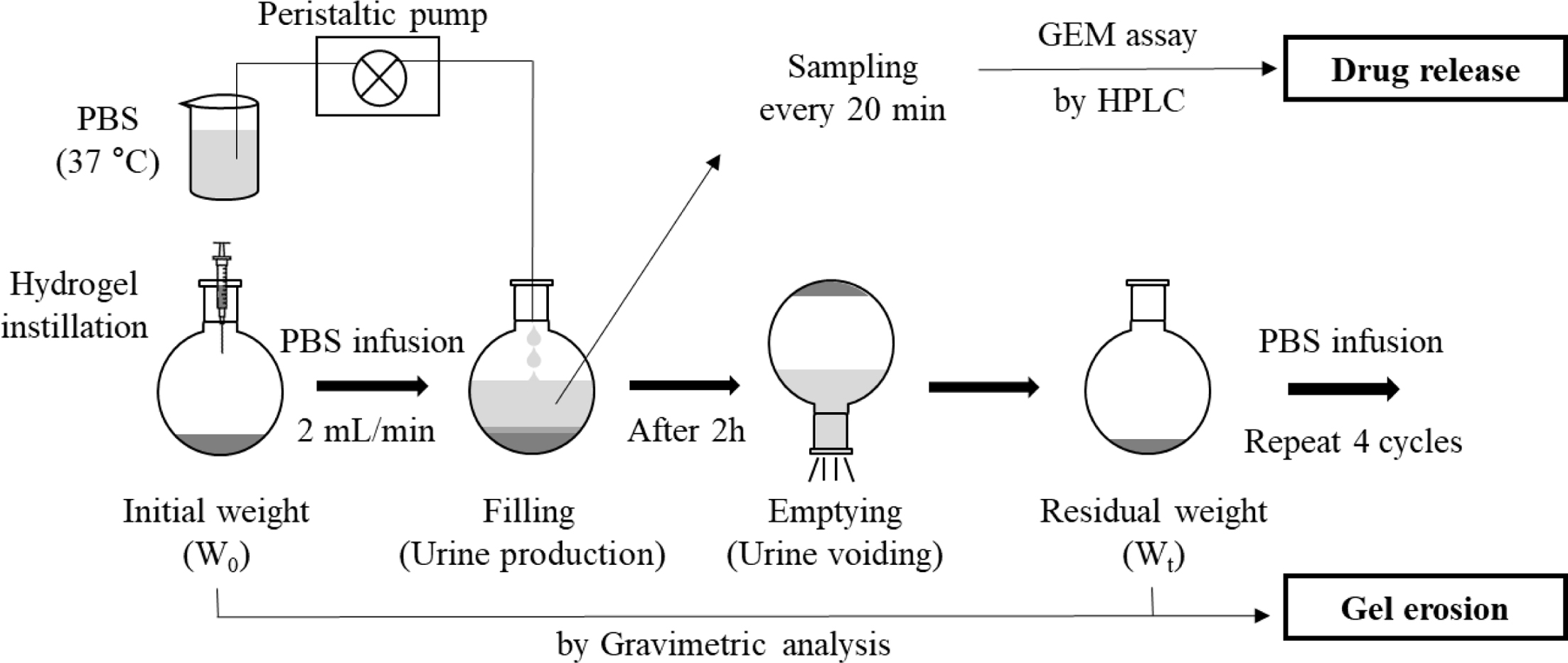
In
Vitro Gel Erosion and Drug Release Study. Based on a
previous study,22 as illustrated in Figure 1, an in vitro
urinary bladder simulation model was adopted to examine gel erosion and drug
release characteristics. Gel erosion was evaluated using the gravimetric method
as previously reported.22 Briefly, 12 mL of hydrogel samples
was instilled into an empty 250 mL round-bottom flask at a constant
temperature of 37 oC using a water bath and the total weight (W0)
was recorded. Subsequently, PBS medium maintained at 37 oC was
added into the flask at a rate of 2 mL/min through a peristaltic pump
(BT100-2J; Longer Precision Pump Co., Ltd., Hebei, China). After peristalsis
for 2 h, the supernatant solution was discarded and the residual weight of the
flask (Wt) was recorded. Fresh PBS medium was infused at the
same rate under the same conditions, and the process was repeated four times.
The remaining fraction of hydrogels (%) was calculated using the following
equation: (Wt/W0) × 100. The
measurements were performed in triplicate.
Simultaneously, drug release from the hydrogel was investigated by
determining the GEM concentration in the PBS solution by high-performance
liquid chromatography (HPLC). Briefly, 0.5 mL of the supernatant solution
was sampled at every 20 min and an equivalent volume of fresh PBS was
replenished to maintain the constant volume of release medium.16
After filtration using a 0.45 μm polyvinylidene fluoride filter (PVDF; Whatman
International Ltd., Maidstone, UK), each sample was subjected to HPLC assay of
GEM. The HPLC system consisted of a pump (W2690/5; Waters Corporation, Milford,
MA, USA), an ultraviolet detector (W2489; Waters Corporation, Milford, MA,
USA), a data station (Empower 3; Waters, Milford, MA, USA), and a ZORBAX ODS
C18 column (4.6 mm ID × 150 mm; Agilent, CA, USA). The mobile phase
was composed of water and acetonitrile (9:1, v/v) and the column temperature
was 25 oC. The flow rate was 1.0 mL/min, and the injection
volume was 20 μL. GEM was detected at a wavelength of 275 nm. The drug release
tests were conducted in triplicate.
Kinetics
Analysis of GEM Release. To investigate the mechanism
of drug release from the prepared hydrogels, different mathematical kinetic
models were employed. The results of the drug release test were fit to the
zero-order kinetics (eq. (1)), first-order kinetics (eq. (2)), Higuchi kinetics
(eq. (3)), Hixson–Crowell equation (eq. (4)), and Ritger–Peppas equation (eq.
(5)) as follows:23,24

where Mt/M∝ is the
fraction of drug released at time t; Mt is the amount
of the drug released at time t; M∝ is the
initial amount of drug in the hydrogel; n is the diffusion exponent; and K0,
K1, KH, KHC, and KRP
are the rate constants for zero-order, first-order, Higuchi,
Hixson–Crowell, and Ritger–Peppas equations, respectively.
Statistical
Analysis. All data are expressed as mean ± standard deviation (n ³ 3). Significant differences were determined using the
Student’s t-test, and the results with a p-value <0.05 were
considered statically significant.
|
Figure 1 Illustration of the in vitro bladder simulation model for drug release and gel erosion study. Filling and emptying cycles repeated
every 2 h. |
Thermo-reversible Property of the Prepared Hydrogels. Various PLX hydrogels were successfully prepared using
the cold technique in the presence or absence of polymeric additives. PLX and
the additives were completely dissolved in water to form a clear solution at
4 oC. Most PLX hydrogels were colorless and transparent, and
the addition of CS and CP resulted in an opaque hydrogel and ALG resulted in a
pale-yellow hydrogel. As shown in Figure 2, all prepared hydrogels exhibited
the thermo-reversible property, that is, free-flowing at 4 oC
but hard-to-flow at 37 oC. Thus, we confirmed that these
behaviors are reversible and that all the selected additives do not alter the
thermo-reversible property of PLX hydrogel.
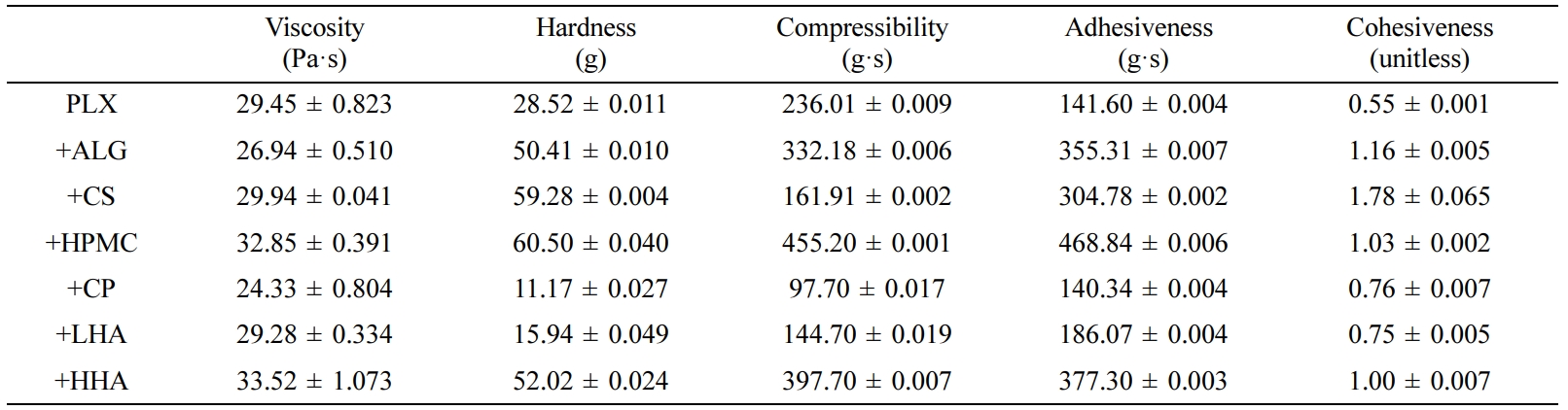
Mechanical
Properties of the Prepared Hydrogels. The viscosity of
hydrogels was measured based on the dependence of non-Newtonian fluids on shear
stress and shear rate, according to the rheological measurements. The viscosity
of the PLX hydrogel was 29.453 Pa·s. The viscosity of different hydrogels decreased
in the following order: HHA > HPMC > CS > plain PLX > LHA > ALG
> CP (Table 1).
The texture analysis could provide an overview of the mechanical
properties of hydrogels, namely, hardness, compressibility, adhesiveness, and
cohesiveness. The hardness and compressibility of the PLX hydrogel were 28.524
g and 236.012 g·s, respectively. The addition of HPMC, ALG, CS, and HHA
increased the hardness to more than 50 g, whereas the addition of CP and
LHA decreased the value to extremely low levels, 11.166 and 15.940 g,
respectively. Similarly, HPMC addition resulted in the highest compressibility
of 455.201 g·s, which was approximately two-fold higher than that of the PLX
hydrogel. The addition of ALG and HHA also increased the compressibility,
whereas the addition of CS, CP, and LHA decreased it to the range of 97.7–161.9
g·s. Overall, CP and LHA resulted in lower hardness and compressibility than
the other additives, indicating the ease of instillation and good spreadability
of hydrogels with these polymers.
The adhesiveness and cohesiveness of the PLX hydrogel were
141.595 g·s and 0.551, respectively. The addition of HPMC significantly
increased the adhesiveness by more than three-fold (468.838 g·s). The other
additives also increased the adhesiveness (g·s) in the following order: HHA
(377.3) > ALG (355.3) > CS (304.8) > LHA (186.1) > CP (140.3).
Similarly, the addition of polymers resulted in a 1.4–3.2-fold increase in
cohesiveness. Specifically, the addition of CS resulted in the highest level cohesiveness
of 1.780, whereas the addition of LHA yielded the lowest (0.746). These results
suggest that HPMC and HHA were preferable among the additives studied.
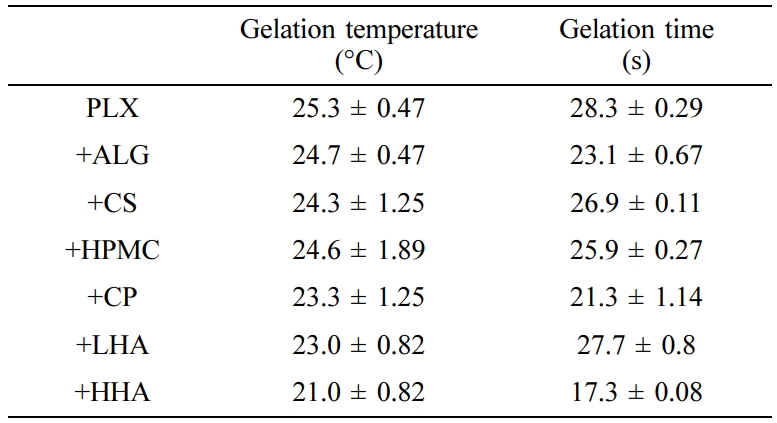
Comparison
of Gel-forming Capacity. The gel-forming
capacity of different hydrogels was compared in terms of gelation temperature
and gelation time (Table 2). These parameters were adopted to estimate the
capacity for long-term retention and rapid gel formation of hydrogel after
instillation into the bladder. The PLX hydrogel presented the highest gelation temperature
of 25.3 oC, whereas HHA-added hydrogel presented the lowest
temperature of 21.0 oC. Thus, in terms of gelation temperature,
all prepared hydrogels should be cooled before instillation.
On the contrary, the PLX hydrogel had the longest gelation time (28.32
s), whereas HHA-added hydrogel had the shortest time (17.28 s). As the gelation
time of the other hydrogels was in the range of 17–30 s, we consider that all
the formulations were injectable to the bladder without any difficulties
including dilution by urine and/or obstruction of catheter needle.
In
Vitro Gel Erosion. The gel erosion
profiles were examined using an in vitro bladder simulation model as
reported previouisly.25,26 In this model, to mimic physiological
urine production and urine voiding, PBS medium was infused into a gel-loaded
flask at 2 mL/min, and after 2 h, the whole medium was discarded; this cycle
was repeated four times, and the gravimetric analysis based on weight loss in
every cycle was applied to obtain the remaining fraction of hydrogels.
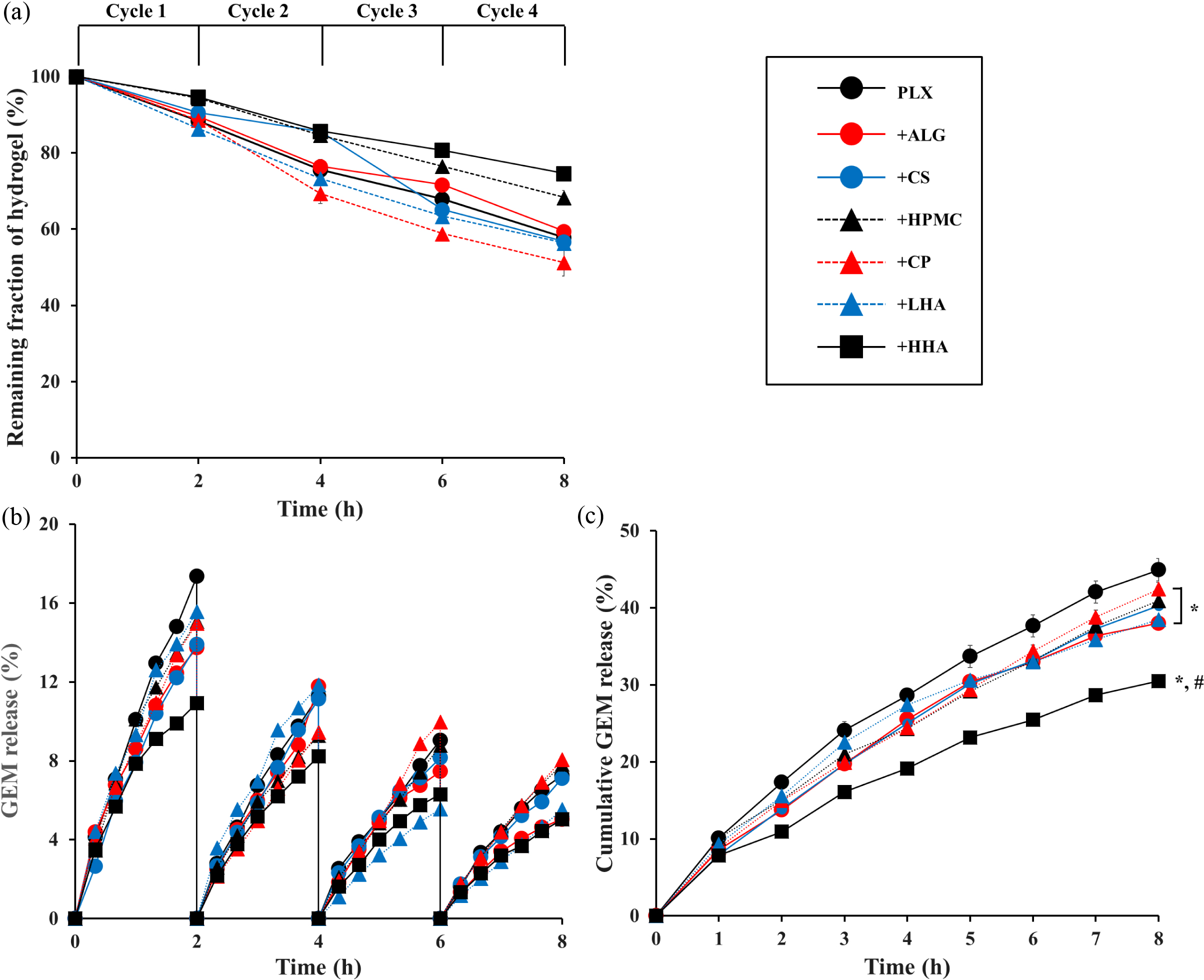
The remaining fraction (%) over four repeated cycles of filling–emptying
was plotted against time (Figure 3(a)). Overall, the remaining fraction of
hydrogels linearly reduced with time, indicating that the erosion followed
zero-order kinetics. After the first cycle (2 h), approximately 5–10% erosion
was observed in most hydrogels, revealing no significant difference between the PLX
hydrogel and polymer-added hydrogels. However, thereafter, differences in the
formulations manifested, with the highest erosion in CP-added hydrogel and the
lowest erosion in HHA-added hydrogel. After four cycles (8 h), erosion in the
remaining fraction (%) of hydrogels decreased in the order of HHA-added (74.6)
> HPMC-added (68.3) > ALG-added (59.4) > PLX (57.8) > CS-added
(56.7) > LHA-added (56.4) > CP-added (51.3).
GEM
Release Profile. The profiles of GEM release were examined every 20 min
over the four repeated cycles. Figure 3(b) represents the plot of the amount of
GEM released versus time in every cycle. In the first cycle (2 h), the
PLX hydrogel showed a higher release than other polymer-added hydrogels, of
17.4%, whereas HHA-added hydrogel showed the most sustained release pattern
(10.9%) and the other polymer-added hydrogels showed an intermediate release pattern
between the above percentages. Furthermore, the degree of GEM release was gradually reduced
with filling–emptying cycles. The degree of drug release for most hydrogels was
maintained in the same order, except for the CP-added and LHA-added hydrogels.
For further comparison, the cycle-based plot was converted to the
cumulative amount plot of GEM release versus time (Figure 3(c)). At 1 h,
no significant differences were found among the hydrogels, showing
approximately 10% GEM release. However, at the end of the first cycle (2 h),
polymer-dependent differences were observed, and the difference increased with
time. After 5 h, compared with the PLX hydrogel, all polymer-added hydrogels
showed a significant difference (p<0.05). In particular, after 3 h,
HHA-added hydrogel showed a significant difference relative to both PLX
hydrogel and other polymer-added hydrogels. After four cycles (8 h), the
cumulative GEM release (%) decreased in the following order: PLX (44.9) >
CP-added (42.4) > HPMC-added (40.9) > CS-added (40.3) > LHA-added
(38.5) > ALG-added (38.0) > HHA-added (30.5). These results suggest that
among the polymeric additives used in this study, HHA was superior in terms of
sustaining the release of GEM.
We prepared thermo-reversible PLX hydrogel formulations to screen for
those with prolonged retention time and sustained drug release in the bladder
cavity. PLX and the selected additives are biocompatible, and they have been
widely used in the pharmaceutical industry owing to their low toxicity. Based on
a previous study, the concentration of PLX was set to 20 w/v%, at which
sensitive sol-to-gel transition and sufficient consistency in the gel state can
be observed.25 Although the content of polymer used in hydrogel
formulations has been shown to vary from 0.1 to 4 w/v%,27 in this
study, the content of polymeric additives was 1 w/v% for relative comparison.
The addition of polymers did not affect the thermo-reversible property of PLX
hydrogel, even though the gelation temperature was slightly reduced. Gelation
temperature is the minimum temperature at which thermo-reversible hydrogel is
in the gel state. To enable phase transition in the bladder cavity after
instillation, the gelation temperature should be below the physiological body
temperature, that is, 37 oC. Gelation time is the time needed
for sol-to-gel transition, which should be within a minute to avoid unexpected
dilution by urine. However, if this gelation time is too short (i.e., within
several seconds), the hydrogel may obstruct the catheter needle during
intravesical instillation. In the present study, the HHA-added hydrogel
presented the lowest gelation temperature (21.0 oC) and
shortest gelation time (17.28 s), which indicate its potential to undergo
gelation after instillation in the bladder and rapidly form a gel. These
results are consistent with those of previous studies.25,28
Here, texture profile and viscosity analyses are proposed as an
appropriate tool to characterize the hydrogel system. Depending on the type of
polymer, the mechanical properties, such as viscosity, hardness,
compressibility, adhesiveness, and cohesiveness, of the hydrogels changed.
Viscosity signifies the molecular movement of viscoelastic materials, and
different viscosities imply differences in the molecular structure, reflecting
intermolecular interaction.29,30 Furthermore, viscosity also
indicates the ease at which a hydrogel can be injected using a syringe, as a
low viscosity is required to reduce the force required to instill.31
Hardness, measured by the peak of force needed to achieve deformation, reflects
the ease of instillation; a low value represents ease of administration.
Compressibility, defined by the work needed to deform the hydrogel during
compression, indicates the ease of spreadability in the inner bladder cavity.
Adhesiveness, measured by the work needed to overcome attractive forces between
the hydrogel surface and probe, is a comparative measure of the adhesive
ability of hydrogels in the bladder cavity and a good indicator of retention in
the bladder.32 Cohesiveness, defined by the ratio of force of the
second deformation to that of the first deformation, is related to the
structural recovery of hydrogel after application in the bladder cavity.33
Selection of appropriate polymer additives is a crucial aspect in
modulating the properties of PLX hydrogel, especially in terms of gel erosion
and drug release. CS is a biodegradable natural polymer.34 By
blending CS with PLX hydrogel, drug release rate can be reduced and residence
time in the administration site can be extended.12,35,36 This is
because of the interpenetration of CS into the PLX gel network, which decreases
the rate of water penetration into the hydrogel. The slower rate of drug
release from CS-added hydrogel is also related to the tortuosity of
microchannels for drug diffusion, which results in a longer time to release the
drug. These characteristics are related to the molecular weight and
concentration of CS.37-39
ALG is a relatively strong mucoadhesive anionic polymer.40 The
physical mixing of ALG with PLX is reported to increase retention time and
sustain drug release from the hydrogel system.11 This phenomenon is
caused by the formation of cross-links between ALG and PLX. The water molecules
may function as a cross-linking agent to create hydrogen bonds between the
carboxyl groups of ALG and the ether group of PLX, which may form a
three-dimensional network. Therefore, this interaction affects the sol–gel
transition temperature and sustained release behavior.13,35 In
addition, the pKa of a drug molecule could be an influencing factor because ALG
is an anionic polymer.41
HPMC is a cellulose derivative. Because of different molecular weights,
they have a wide range of viscosity, adsorptive activities, and osmotic
properties. Because of these characteristics, HPMC is used as a component of
various gels including dermatological and ophthalmological gels.42
The sustained release property of HPMC-added hydrogel has been explained by a
previous study,43 which described that the viscosity of hydrogels
increased with the use of HPMC owing to its ability to bind the poly EO chains,
increasing the entanglement of the adjacent molecules. Thereby, the polymer
additives decrease the initial burst and prolong drug release. Similarly, Zhang
et al. reported that HPMC swells in solution and forms disordered
physical networks. Thus, the extensive hydrogen bonds and the molecule
entanglement could form tightly orientated hydrogel structure, resulting in
delayed drug diffusion and gel dissolution.44
CP, also known as a carbomer, is an acrylic acid derivative, which is
widely used as a gel-forming agent in soft medicinal formulations. There are
various brands of CP, classified according to the rate of gel formation,
transparency of gels, and ability to suspend and emulsify.42
Moreover, they have excellent mucoadhesive properties in ocular delivery and
sustained drug release profile when mixed with PLX.16 The reasons of
the sustained drug release profile of CP-added hydrogel have been explained by
a previous study, which reported that CP can form a polymer complex with PLX by
hydrogen bonding, subsequently reinforcing the network structure and slow
relaxation and hydration of the polymer matrix.45
HA is a natural biodegradable polysaccharide.46 It is used
alone or in combination with others and is mainly used for joint lubrication
and ocular delivery.47 It is known that HA, when mixed with PLX
hydrogel, increases gel strength and decreases gel network porosity. In
addition, Mayol et al. reported that HAs of different molecular weights
vary in rheological behavior when mixed with PLX hydrogel.48 The
sustained release property of the HA molecules is because they can interact
with PLX micelles via hydrogen bonding, thus reinforcing the structure of
micelles and mechanical properties of hydrogel.48 Moreover, the
possibility of interaction between HHA random coils and PLX micelles increases
aggregation relative to that by PLX-only micelles or PLX-LHA micelles.48
The micelle movement is increasingly hampered as PLX gelation progresses. A
similar study showed that the addition of HHA resulted in a sustained release,
while preventing the initial burst.49 The reason for this behavior
is ascribed to the closely packed inter-micellar structure by the addition of
HHA. Furthermore, the highly packed supramolecular structure reduces the
diffusion coefficients of HHA-added hydrogel, which could result in prolonged
drug release. These findings are consistent with our results, in which
HHA-added hydrogel showed better sustained GEM release behavior than the other
polymer-added hydrogels.
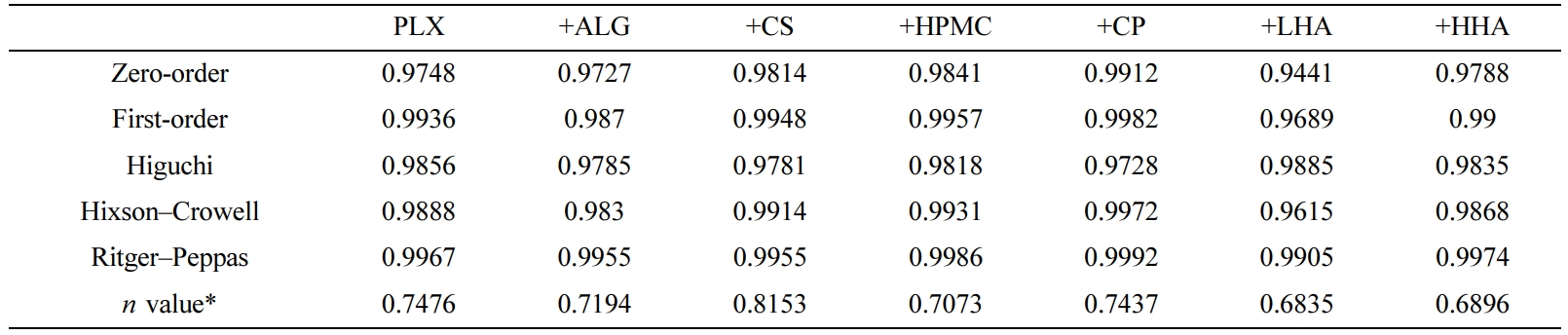
Mathematical kinetic models are an important tool to evaluate the drug
release process. They can be used to evaluate some important physical
parameters and have recourse for model fitting experimental release data.50
By curve fitting to the different kinetics, such as the zero-order,
first-order, Higuchi, Hixson–Crowell, and Ritger–Peppas equations, correlation
coefficient (R2) of all formulations were obtained. All R2
values were greater than 0.9 (Table 3), indicating a good linearity. Among
various kinetic models, the Ritger–Peppas model was the best fit with the
highest R2 value. Specifically, this model is useful to
characterize polymeric systems, in which the release mechanism is not fully
understood or complex.50 In addition, the Ritger–Peppas model
provides the value of diffusion exponent (n), which better determines
the mechanism of a drug diffusion from a matrix system whether the diffusion is
Fickian or non-Fickian. As shown in Table 3, all formulations presented values
in the range of 0.6835–0.8153, indicating non-Fickian diffusion. This type of
anomalous release is governed by swelling and diffusion. The slow rearrangement
of polymeric chains (swelling and finally erosion) and simultaneous diffusion
induce the time-dependent anomalous effects.51 Therefore, all
additives may follow the erosion–diffusion mechanism. Thus, both hydrogel
erosion and GEM diffusion are important processes in GEM release from the
hydrogels tested in this study.
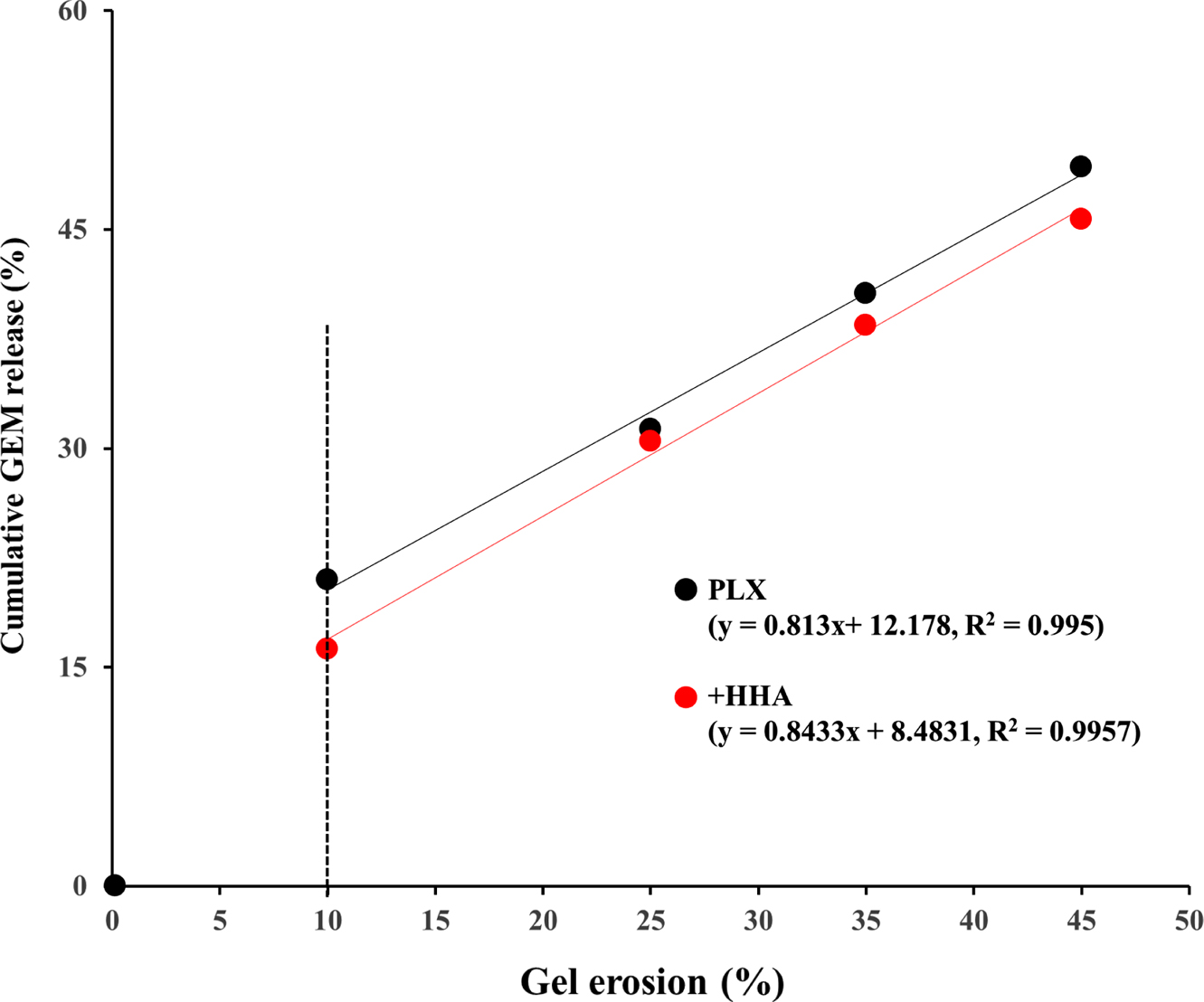
Furthermore, we investigated the correlations between gel erosion and
drug release. The PLX hydrogel and HHA-added hydrogel were selected as
representatives for the analysis, and the cumulative amount of GEM release (%)
was plotted against gel erosion (%). For plotting, the values at specific time
points were obtained using the regression equations of the Ritger–Peppas model
(GEM release) and zero-order kinetics (gel erosion). Both PLX and HHA-added
hydrogels showed a similar pattern (Figure 4), indicating negligible influence
of polymer addition on this correlation. Until 10% erosion, there was an
initial burst, which can be attributed to the leakage of drug during gel
formation and/or rapid diffusion of drug molecules adjacent to bulk front. The
addition of HHA decreased the level of initial burst. Except at this period,
correlations between gel erosion and GEM release were well-established for both
hydrogels, with the R2 values of 0.9950 and 0.9957 for PLX
hydrogel and HHA-added hydrogel, respectively. Moreover, the slope of
regression equations was close to unity, implying GEM release proportional to
gel erosion. Several studies have described the erosion-controlled release
kinetics of hydrogels.26 Other factors such as molecular size and
hydrophilicity are also important. If a drug molecule is hydrophobic, drug
release is mainly governed by gel erosion. However, as GEM, with a molecular
weight of 263 Da, is water soluble, drug release is governed by both gel
erosion and molecular diffusion. Therefore, we expect that these hydrogels,
preferably HHA-added hydrogels, are good candidates for developing an in-situ
gelling system to increase drug exposure in bladder cancer treatment.

|
Figure 2 Observation of thermo-sensitive phase transition. |
|
Figure 3 Results of in vitro gel erosion and gemcitabine (GEM) release. (a) Plot of remaining fraction of hydrogel (%) versus time for four
repeated cycles. (b) Plot of GEM release (%) versus time for every cycle. (c) Plot of cumulative GEM release (%) versus time. Significantly
different at p<0.05: *versus PLX hydrogel; #
versus other polymer-added hydrogel. Values are presented as mean ± SD (n=3). |
|
Figure 4 Relationship between cumulative GEM release (%) and
gel erosion (%) for plain PLX and HHA-added hydrogel. Values at
specific time points were calculated using the regression equations
of the Ritger–Peppas model (GEM release) and zero-order kinetics
(gel erosion). |
|
Table 1 Mechanical Properties of Various Hydrogels |
Values are presented as mean ± SD (n = 3). |
|
Table 2 Gel-forming Capacity of Various Hydrogels
Gelation temperature
(°C)
Gelation ti |
Values are presented as mean ± SD (n = 3). |
|
Table 3 Comparison of Correlation Coefficients (R2
) in Various Mathematical Kinetic Models |
*Diffusion exponent related to the Ritger–Peppas model. |
PLX-based hydrogels with or without polymer additives were successfully
formulated while maintaining its thermo-reversible property. With excellent
gel-forming capacity, the hydrogels revealed acceptable mechanical properties
in terms of viscosity, gel strength, and adhesiveness. Drug release was
diffusion and erosion controlled. Specifically, HHA-added hydrogel is a
promising system for intravesical instillation in the treatment of bladder
cancer. However, further in vivo assessments are still needed.
- 1. S. Antoni, J. Ferlay, I. Soerjomataram, A. Znaor, A. Jemal, and F. Bray, Eur. Urol., 71, 96 (2017).
-
- 2. H. Rubben, H. Lutzeyer, N. Fischer, F. Deutz, W. Lagrange, and G. Giani, J. Urol., 139, 283 (1988).
-
- 3. D. S. Kaufman, W. U. Shipley, and A. S. Feldman, Lancet, 374, 239 (2009).
-
- 4. M. K. Nguyen and D. S. Lee, Macromol. Biosci., 10, 563 (2010).
-
- 5. A. S. Hoffman, Adv. Drug Deliv. Rev., 54, 3 (2012).
- 6. C. He, S. W. Kim, and D. S. Lee, J. Control. Release, 127, 189 (2008).
-
- 7. L. Yu and J. Ding, Chem. Soc. Rev., 37, 1473 (2008).
-
- 8. A. V. Kabanov, E. V. Batrakova, and V. Y. Alakhov, J. Control. Release, 82, 189 (2002).
-
- 9. J. Juhasz, V. Lenaerts, P. Raymond, and H. Ong, Biomaterials, 10, 265 (1989).
-
- 10. G. Dumortier, J. L. Grossiord, F. Agnely, and J. C. Chaumeil, Pharm. Res., 23, 2709 (2006).
-
- 11. H. Abdeltawab, D. Scirskis, and M. Sharma, Expert. Opin. Drug Deliv., 17, 495 (2020).
-
- 12. A. C. S. Akkari, J. Z. B. Papini, G. K. Garcia, K. K. Margareth, D. Franco, L. P. Cavalcanti, A. Gasperini, M. I. Alkschbirs, F. Yokaichyia, E. D. Paula, G. R. Tofoli, and D. R. Araujo, Mater. Sci. Eng. C, 68, 299 (2016).
-
- 13. H. R. R. Lin, K. C. Sung, and W. J. J. Vong, Biomacromolecules, 5, 2358 (2004).
-
- 14. L. Mayol, F. Quaglia, A. Borzacchiello, L. Ambrosio, and M. I. L. Rotonda, Eur. J. Pharm. Biopharm., 70, 199 (2008).
-
- 15. T. G. Yarnykh, E. V. Tolochko, and V. N. Chushenko, J. Pharm. Chem., 44, 551 (2011).
-
- 16. H. Qi, W. Chen, C. Huang, L. Li, C. Chen, W. Li, and C. Wu, Int. J. Pharm., 337, 178 (2007).
-
- 17. I. R. Schmolka, J. Biomed. Mater. Res., 6, 571 (1972).
-
- 18. K. S. Sandhu and N. Singh, J. Food Chem., 101, 1499 (2007).
-
- 19. A. Kluk and M. Sznitowska, Int. J. Pharm., 460, 228 (2014).
-
- 20. C. Ju, J. Sun, P. Zi, X. Jin, and C. Zhan, J. Pharm. Sci., 102, 2707 (2013).
-
- 21. Q. F. Dang, J. Q. Yan, J. J. Li, X. J. Cheng, C. S. Liu, and X. G. Chen, Carbohydr. Polym., 83, 171 (2011).
-
- 22. T. Lin, J. Wu, X. Zhao, H. Lian, A. Yuan, X. Tang, S. Zhao, H. Guo, and Y. Hu, J. Pharm. Sci., 103, 927 (2014).
-
- 23. L. Mayol, M. Biondi, F. Quaglia, S. Fusco, A. Borzacchiello, L. Ambrosio, and L. Rotonda, Biomacromolecules, 12, 28 (2011).
-
- 24. N. Ahuja, O. P. Katare, and B. Singh, Eur. J. Pharm. Biopharm., 65, 26 (2007).
-
- 25. S. H. Kim, S. R. Kim, H. Y. Yoon, I. H. Chang, Y. M. Whang, M. J. Cho, M. J. Kim, S. J. Lee, and Y. W. Choi, Korean J. Urol. Oncol., 15, 178 (2017).
-
- 26. D. Grennwood and F. O’Grady, J. Antimicrob. Chemother., 4, 113 (1978).
-
- 27. E. Giuliano, D. Paolino, M. Fresta, and D. Cosco, Pharmaceutics, 10, 159 (2018).
-
- 28. M. Dewan, G. Sarkar, M. Bhowmik, B. Das, A. K. Chattoapadhyay, D. Rana, and D. Chattopadhyay, Int. J. Biol. Macromol., 102, 258 (2017).
-
- 29. J. M. Quintana, A. N. Califano, N. E. Zaritzky, P. Partal, and J. M. Franco, J. Texture. Stud., 33, 215 (2002).
-
- 30. J. Desbrieres, Biomacromolecules, 3, 342 (2002).
-
- 31. C. M. Chang and R. Bodmeier, Int. J. Pharm., 173, 51 (1988).
-
- 32. J. Hurler, A. Engesland, B. P. Kermany, and N. S. Basnet, J. Appl. Polym. Sci., 125, 180 (2012).
-
- 33. G. Calixto, A. C. Yoshii, H. R. Silva, B. S. F. Cury, and M. Chorilli, Pharm. Dev. Technol., 20, 490 (2014).
-
- 34. H. Gupta, A. Sharma, and B. Shrivastava, Asian J. Pharm., 3, 94 (2009).
-
- 35. V. Sridhar, S. Wairkar, R. Gaud, A. Bajaj, and P. Meshram, J. Drug. Target., 26, 150 (2018).
-
- 36. A. Y. Sherif, G. M. Mahrous, and F. K. Alanazi, Saudi Pharm. J., 26, 845 (2018).
-
- 37. F. Tuğcu-Demiröz, Marmara Pharm. J., 21, 762 (2017).
-
- 38. T. Ur-Rehman, S. Tavelin, and G. Gröbner, Int. J. Pharm., 409, 19 (2011).
-
- 39. H. J. Cho, P. Balakrishnan, E. K. Park, K. W. Song, S. S. Hong, T. Y. Jang, K. S. Kim, S. J. Chung, C. K. Shim, and D. D. Kim, J. Pharm. Sci., 100, 681 (2011).
-
- 40. N. K. Sachan, S. Pushkar, A. Jha, and A. Bhattcharya, J. Pharm. Res., 2, 1191 (2009).
- 41. H. U. Lin, K. C. Sung, and W. J. Vong, Biomacromolecules, 5, 2358 (2004).
-
- 42. M. N. Anurova, E. O. Bakhrushina, and N. B. Demia, J. Pharm. Chem., 49, 627 (2015).
-
- 43. Z. G. Yu, Z. X. Geng, T. F. Liu, and F. Jiang, J. Vet. Pharmacol. Therap., 38, 271 (2014).
-
- 44. L. Zhang, D. L. Parsons, C. Navarre, and U. B. Kompella, J. Control. Release, 85, 73 (2002).
-
- 45. K. R. Lee, E. J. Kim, S. W. Seo, and H. K. Choi, Arch. Pharm. Res., 31, 1023 (2008).
-
- 46. K. Y. Cho, T. W. Chung, B. C. Kim, M. K. Kim, J. H. Lee, W. R. Wee, and C. S. Cho, Int. J. Pharm., 260, 83 (2003).
- 47. E. J. Strauss, J. A. Hart, M. D. Miller, R. D. Altman, and J. E. Rosen, Am. J. Sports. Med., 37, 1636 (2009).
-
- 48. L. Mayol, F. Quaglia, A. Borzacchiello, L. Ambrosio, and M. I. Rotonda, Eur. J. Pharm. Biopharm., 70, 199 (2008).
-
- 49. M. H. M. Nascimento, M. K. K. D. Franco, F. Yokaichyia, E. de Paula, C. B. Lombello, and D. R. de Araujo, Int. J. Biol. Macromol., 111, 1245 (2018).
-
- 50. N. A. Peppas and B. Narasimhan, J. Control. Release, 190, 75 (2014).
-
- 51. Bruschi and M. Luciano, Strategiesto Modify the Drug Release from Pharmaceutical Systems, WoodheadPublishing, 2015.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2020; 44(6): 817-826
Published online Nov 25, 2020
- 10.7317/pk.2020.44.6.817
- Received on Jun 16, 2020
- Revised on Jul 28, 2020
- Accepted on Aug 4, 2020
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Wook Choi
-
College of Pharmacy, Chung-Ang University, 84 Heuksuk-ro, Dongjak-gu, Seoul 06974, Korea
- E-mail: ywchoi@cau.ac.kr
- ORCID:
0000-0003-2431-3995
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr