






- Polymer Solar Cells based on a Furan-containing Asymmetric Nonfullerene Acceptor
Seunggyun Hong, Chang Eun Song*,**, and Eunhee Lim†

Department of Chemistry, Kyonggi University, 154-42 Gwanggyosan-ro, Yeongtong-gu, Suwon 16227, Korea
*Korea Research Institute of Chemical Technology (KRICT), 141 Gajeongro, Yuseong-gu, Daejeon 34114, Korea
**University of Science and Technology (UST), Daejeon 34113, Korea- 퓨란이 도입된 비대칭 비풀러렌계 어셉터를 기반으로 하는 고분자 태양전지
홍승균 · 송창은*,** · 임은희†
경기대학교 화학과, *한국화학연구원, **한국과학기술연합대학원대학교(UST)
An asymmetric nonfullerene
small molecule, TF-ORH, containing a nonfused electron-donating core of
thiophene, furan and electron-withdrawing octylrhodanine (ORH) end groups was
designed as an acceptor for polymer solar cells (PSCs). TF-ORH was synthesized via
Knoevenagel condensation and the Suzuki coupling reaction. TF-ORH exhibited
sufficient thermal stability and solubility for device fabrication. Compared to
solution, TF-ORH film showed a red-shifted UV–Vis absorption with an absorption
maximum at 596 nm (J-aggregation), which is complementary with that of
the low bandgap polymer donor of
poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b']dithiophene-co-3-fluorothieno[3,4-b]thiophene-2-carboxylate]
(PTB7-Th). The introduction of furan elevated the molecular orbital energy
levels of TF-ORH compared to the thiophene analog, T2-ORH. Finally, a PSC
fabricated using a PTB7-Th:TF-ORH blend film as the active layer exhibited a power
conversion efficiency of 1.21% with an open-circuit voltage of 0.88 V and a small energy loss of
0.70 eV.
고분자 태양전지의 어셉터로 비접합된 전자 주는 thiophene-furan 중심과 전자 끄는 옥틸로다닌 말단 그룹으로 이루어진 비대칭 비풀러렌 단분자 TF-ORH가 설계되었다. TF-ORH은 Knoevenagel 축합과 스즈끼 짝지음 반응을 통해 합성되었다.
TF-ORH는 소자 제작에 충분한 열 안정성 및 용해도를 나타냈다. 용액과 비교하여, TF-ORH 필름은 596 nm에서 최대 흡수를 가지는
장파장 이동된(J-aggregation) UV-Vis 흡수를 나타내었고, 이는 저밴드갭 고분자 도너인 PTB7-Th의 특성과 상호보완적이다. 퓨란의 도입으로 thiophene 유사체인 T2-ORH와 비교하여 분자 궤도 에너지 준위들은 높아졌다. 활성층으로서 PTB7-Th:TF-ORH 블렌드 필름을 사용하여 제작된 고분자 태양전지 소자는 0.88 V의 개방전압과 0.70 eV의 낮은 에너지
손실로 1.21%의 전력 변환 효율을 나타냈다.
A power conversion efficiency of 1.21% was achieved in
polymer solar cells based on low bandgap polymer donor, PTB7-Th, and an
nonfused asymmetric nonfullerene acceptor, TF-ORH.
Keywords: organic solar cell, polymer solar cell, non-fullerene acceptor, asymmetrical structure
This work was financially supported by the Korea Institute
of Energy Technology Evaluation & Planning (KETEP) and the Ministry of
Trade, Industry & Energy (No. 20173010012960) and by the National Research
Foundation of Korea (NRF) Grant funded by the Ministry of Science & ICT
(NRF-2019R1A2C1003679) of the Republic of Korea. This work was supported by
Kyonggi University’s Graduate Research Assistantship 2019.
Information is available regarding synthesis, 1H and 13C
NMR spectra, physical measurement, and device fabrication.
PK_2020_044_05_741_Supporting_Information_template.pdf (523 kb)
Supplementary Information
Polymer solar cells (PSCs) have considered as a renewable energy source
owing to their advantages, which include low weight, flexibility, ease of processability,
and low cost.1-5 Much effort has been devoted to exploiting new
donor and acceptor materials as well as to optimize device fabrication. From
the standpoint of material synthesis, in recent years there has been a shift
from using fullerene derivatives, such as [6,6]-phenyl-C61-butyric
acid methyl ester (PC60BM), to using non-fullerene acceptors (NFAs)
as acceptor materials. The use of NFAs solves key intrinsic drawbacks of
fullerenes, which include difficult chemical structure modification, weak UV–vis
absorption, fixed energy levels, low stability, and large energy losses (Elosss).
The relatively easy synthesis of NFAs with various molecular structures enables
the control of their physical properties, allowing the development of various
electron acceptors with desirable properties. These acceptors can be used for
highly efficient and stable PSCs for future practical applications.6
Acceptor–donor–acceptor (A–D–A)-type NFAs have been considered promising
candidates for high efficiency PSC acceptors.7,8 In particular,
remarkable power conversion efficiencies (PCEs) more than 15% have been
realized in devices using ladder-type fused ring electron acceptors (FREAs)
with large π-conjugated cores including indacenodithiophene and
indacenodithieno[3,2-b]thiophene.9,10 Two representative
A–D–A-structured low bandgap acceptors are IT-4F and Y6, both of which provide
high efficiencies of >14% when combined with various wide bandgap polymer
donors.11 Various synthetic strategies have been used to adjust the
chemical structure of NFAs with a view to maximizing PSC efficiency, including
side-chain engineering and introduction of electron-rich or electron-poor
substituents into the backbones. Recently, the development of asymmetric NFAs
has made significant progress toward higher PCEs, with asymmetric NFAs showing
superior performance to their symmetric counterparts.12-15 Various
NFAs with asymmetric cores, side-chains, and terminal groups have been
designed;16-18 however, most successful asymmetric NFAs have been based
on asymmetric FREA cores. Possible reasons for the superiority of asymmetric
NFAs are that their structures may give rise to stronger intermolecular
interactions, higher electron mobility in the film, and higher charge carrier
transport. For greater detail, refer to recent reviews on the structure and
characteristics of high-efficiency asymmetric acceptors.16
Meanwhile, thiophene analogs, such as selenophene,19-21 thiazole,22-24
and furan,25-27 have been utilized to develop π-conjugated
polymers and small molecules. Some of these thiophene analogs of furan or
selenophene have recently been introduced into asymmetric NFAs.28,29
In this study, we applied the “asymmetric core” strategy to our
previously reported A–D–A-type NFA, T2-ORH, containing a nonfused bithiophene
core as an electron-donating (D) unit and octylrhodanine end groups as
electron-accepting (A) units.30 In the current work, one thiophene
of the bithiophene core of T2-ORH was replaced with a furan moiety, resulting
in a simple asymmetric NFA, TF-ORH, based on a nonfused thiophene-furan core.
The synthesis and physical properties, including thermal, optical, and
electrochemical properties, of TF-ORH were investigated in detail and a PSC was
fabricated using TF-ORH and the low bandgap polymer donor
poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4, 5-b']dithiophene-co-3-fluorothieno[3,4-b]thiophene-2-carboxylate] (PTB7-Th).
Synthesis. The syntheses of
octylrhodanine (ORH) and B-T-ORH (Scheme 1) were described in our previous
literatures.18,30 Br-F-ORH and TF-ORH were synthesized via
Suzuki coupling and Knoevenagel condensation reaction, respectively.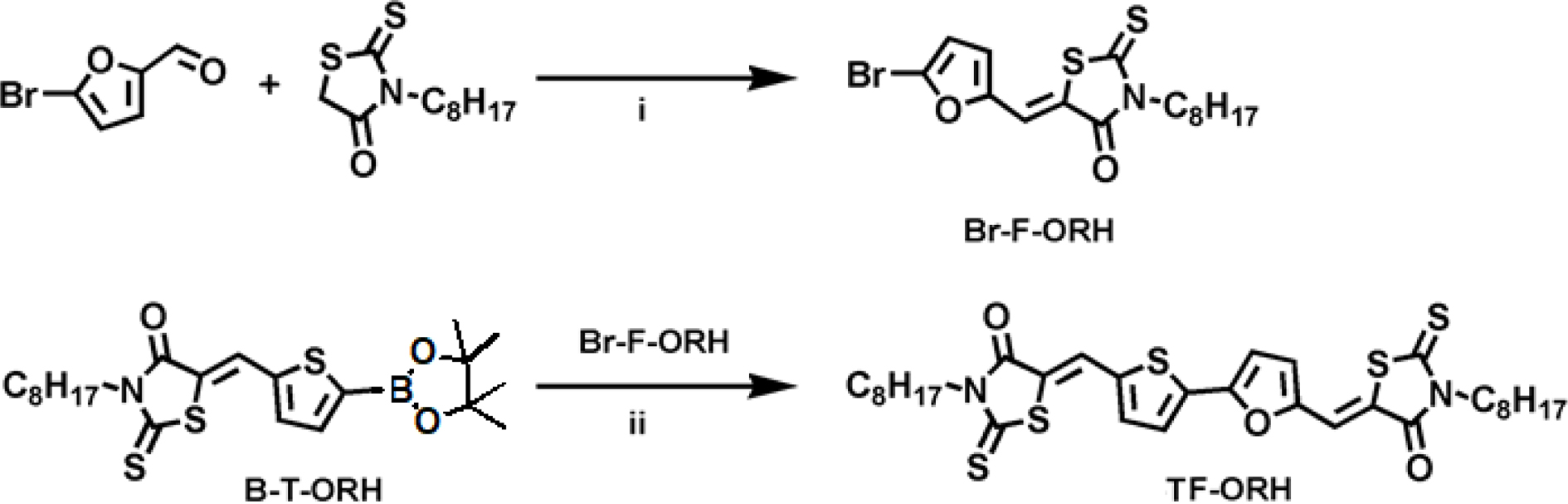
Scheme 1. The synthesis of TF-ORH. (i) Piperidine, chloroform, 5 min; (ii) Pd2(dba)3, K3PO4 (aq, 1 M), THF, reflux, N2, 4 h.
Synthesis
of Br-F-ORH: 5-Bromo-2-furaldehyde (0.84 g, 4.8 mmol) was dissolved in
anhydrous chloroform (30 mL), and a few drops of piperidine and ORH
(2.94 g, 12.0 mmol) were added. After refluxed and stirred for
5 min under an N2 atmosphere, the reaction mixture was
extracted with dichloromethane, washed with water, and dried over MgSO4.
After purification by column chromatography (dichloromethane:
hexane = 1:1), the compound Br-F-ORH was obtained as a yellow solid
(1.39 g, 72% yield). 1H NMR (400 MHz, CDCl3): d (ppm) 7.35 (s, 1H), 6.76 (d, J = 3.6 Hz, 1H), 6.52
(d, J = 3.6, 1H), 4.09 (t, J = 7.7 Hz, 2H), 1.69 (m, 2H), 1.27
(m, 10H), 0.88 (t, J = 6.9 Hz, 3H).
Synthesis
of TF-ORH: Degassed H2O solution of potassium phosphate
tribasic (K3PO4, 1 M, 7.2 mL) was added to 30 mL of
tetrahydrofuran (THF) solution of Br-F-ORH (0.95 g, 2.36 mmol),
B-T-ORH (1.10 g, 2.36 mmol), tri-tert-butylphosphonium
tetrafluoroborate (P(t-Bu)3·HBF4, 0.041 g,
0.14 mmol), and tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3),
0.065 g, 0.07 mmol). After refluxed and stirred for 4 h under an N2
atmosphere, the reaction mixture was extracted with dichloromethane, washed
with water, and dried over MgSO4. After recrystallized by methanol
and purified by column chromatography (chloroform:hexane = 5:1), the
final product of TF-ORH was obtained as a purple solid (0.25 g, 16%
yield). 1H NMR (400 MHz, CDCl3): d (ppm) 7.85 (d, 0.5 Hz, 1H), 7.53 (d, J = 4.0 Hz,
1H), 7.45 (s, 1H), 7.41 (d, J = 4.0, 0.5 Hz, 1H), 6.93 (d, J =
3.8 Hz, 1H) 6.84 (d, J = 3.8 Hz, 1H), 4.12 (m, 4H), 1.71 (m, 4H), 1.27
(m, 20H), 0.88 (t, J = 6.9 Hz, 6H). 13C NMR (126 MHz, CDCl3):
d (ppm) 193.99, 191.88, 167.64, 167.62, 152.39, 150.57,
138.88, 138.36, 134.94, 126.66, 124.38, 122.44, 122.11, 121.05, 116.85, 111.39,
45.18, 44.97, 31.99, 29.35, 27.24, 27.20, 27.00, 22.85, 14.31. Anal. Calc. for
C32H40N2O3S5: C, 58.15;
H, 6.10; N, 4.24; S, 24.26. Found: C, 55.96; H, 5.88; N, 4.30; S, 23.88.
Synthesis
and Thermal Properties. The nonfused A–D–A-type NFA, TF-ORH, was designed
to have an asymmetric electron-donating core (TF) consisting of thiophene and
furan, along with electron-withdrawing rhodanine (RH) groups at both ends, thus
enabling TF-ORH to function as an acceptor in PSCs. The intermediate Br-F-ORH
was synthesized through Knoevenagel condensation of bromo-2-furaldehyde and ORH
in the presence of piperidine catalyst. TF-ORH was then obtained via the
Suzuki coupling reaction of B-T-ORH and Br-F-ORH using a Pd2(dba)3
catalyst. The synthetic procedures are shown in Scheme 1. The purity of TF-ORH
was verified by 1H and 13C NMR spectroscopy and
elementary analyses (Figures S1-S2). TF-ORH was soluble in chloroform at a
concentration sufficient for device fabrication.
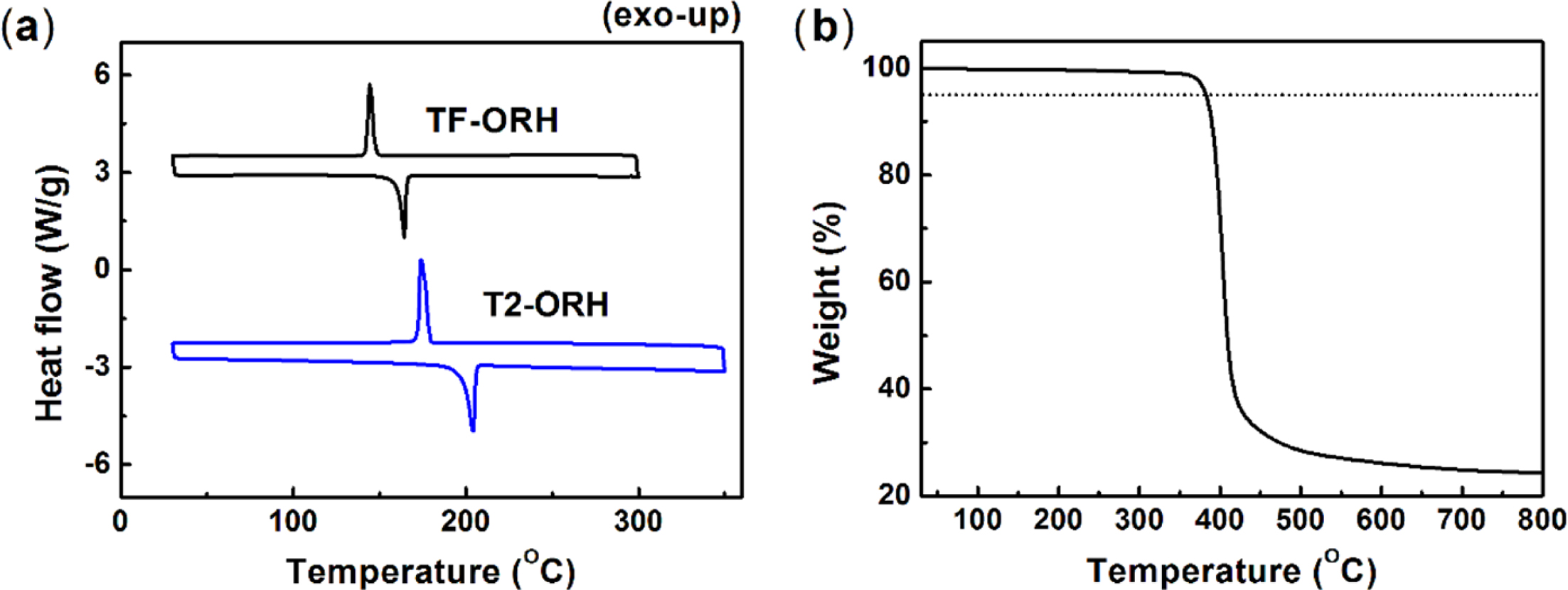
Thermogravimetric analysis (TGA) and differential scanning calorimetry
(DSC) were used to investigate the thermal properties of TF-ORH (Figure 1 and
Table 1). TF-ORH showed a 5% loss of its initial weight at 383 oC,
which was almost the same as T2-ORH,30 indicating that its thermal
stability is sufficient for device applications. In the DSC experiments, TF-ORH
exhibited melting and crystallization temperatures of 164 and 145 oC,
respectively, with both of these phase transition temperatures being higher
than the corresponding values for T2-ORH (204 and 175 oC,
respectively).30 The introduction of furan also lowered the enthalpy
changes of melting (ΔHm) and
crystallization (ΔHcryst) from –70.0 and
68.8 J g–1, respectively, for T2-ORH18 to –45.3 and
44.6 J g–1, respectively, for TF-ORH. Therefore, the asymmetric
configuration created by introducing the furan moiety effectively lowered the
molecular aggregation of the TF-ORH film, which is consistent with the higher
solubility of TF-ORH than that of T2-ORH.
Optical
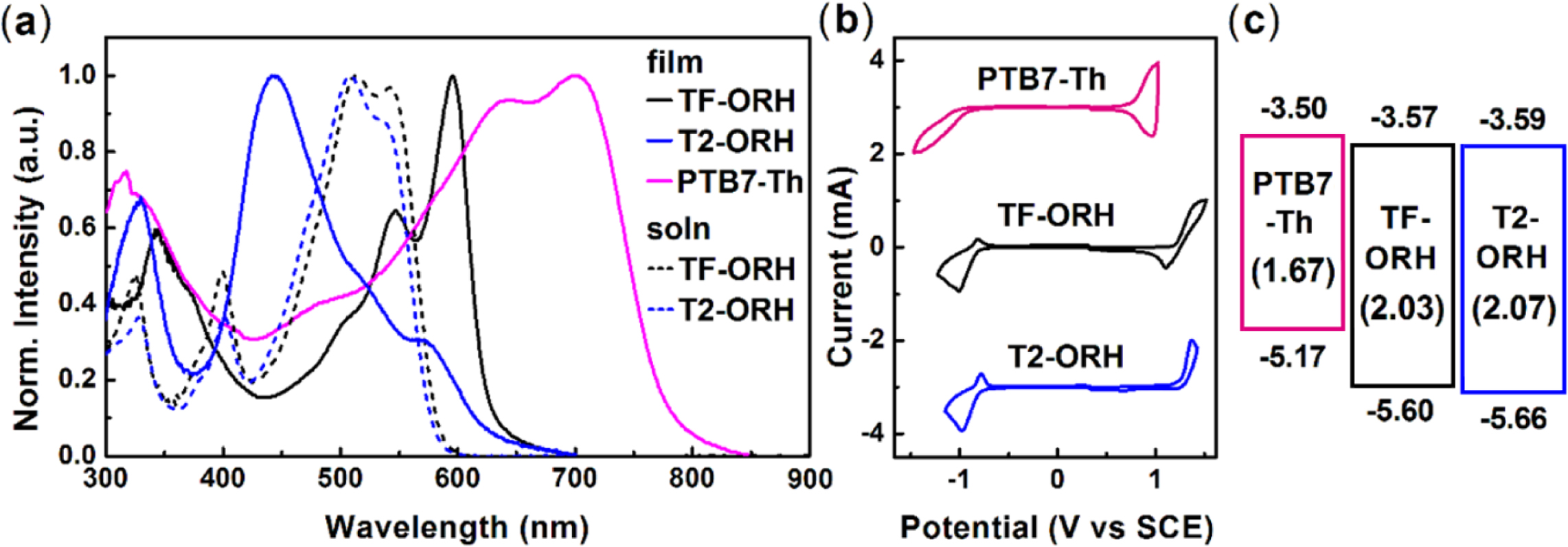
and Electrochemical Properties. The UV–Vis absorption spectra of TF-ORH were measured and compared with
those of T2-ORH and PTB7-Th (Figure 2(a)). The solution UV–Vis spectrum of
TF-ORH showed an absorption maximum (λmax) at 513 nm, almost
the same as the λmax of T2-ORH. The
relatively short conjugation length (i.e., two nonfused aromatic rings
flanked by two RHs) of TF-ORH and T2-ORH resulted in a UV–Vis absorption at
wavelengths below 600 nm. The λmax of the TF-ORH film
was 596 nm, which was red-shifted compared to the solution value, suggesting J-type
aggregation. In contrary, the absorption of the as-cast T2-ORH film (λmax = 445 nm) was blue-shifted relative to
the solution value, consistent with H-aggregation.30 In
addition, the absorption of the T2-ORH film was red-shifted by annealing;30
however, no absorption change was observed in the TF-ORH film (Figure S3). These
findings suggest that the substitution of thiophene with furan changed the
molecular packing behavior of the films The absorption spectrum of TF-ORH was
complementary with that of the low bandgap polymer, PTB7-Th, which is
beneficial for PSC applications. The optical energy bandgaps (Eg,opt)-calculated
using the equation Eg,opt = 1240/λonset (eV), where λonset is the absorption onset of the film-were 1.98 and
1.58 eV for TF-ORH and PTB7-Th, respectively.
The electrochemical properties were determined using cyclic voltammetry
(CV). The films were prepared by dip-coating the small molecule solution onto
the Pt working electrode, and the measurements were calibrated using the
ferrocenium (Fc+)/ferrocene (Fc) redox value of –4.8 eV as an external
reference. The highest occupied molecular orbital (HOMO) and lowest unoccupied
molecular orbital (LUMO) energy levels were estimated according to the
empirical relationship EHOMO = –(Eonset,ox – E1/2,Fc
+ 4.8)
eV and ELUMO = –(Eonset,red – E1/2,Fc + 4.8)
eV, where Eonset,ox, Eonset,red, and E1/2,Fc
are the onset potentials of oxidation and reduction, and half-wave potential of
Fc/Fc+ couple, respectively, assuming that the energy level of Fc is
4.8 eV below the vacuum level.31,32 The HOMO and LUMO energy levels
of TF-ORH were calculated to be –5.60 and –3.57 eV, respectively, resulting in
an electrochemical energy bandgap (Eg,CV) of 2.03 eV. The
energy diagrams of TF-ORH, T2-ORH, PTB7-Th are shown in Figure 2. The Eg,CV
of TF-ORH was slightly smaller than that of T2-ORH (2.07 eV). Compared to the
energy levels of T2-ORH, the introduction of furan elevated the energy levels
of the small molecule. A slightly stronger elevating effect of furan on the
HOMO levels than on the LUMO levels resulted in the relatively smaller Eg,CV
of TF-ORH. In addition, when the polymer donor is PTB7-Th (EHOMO = –5.17 eV and ELUMO = –3.50
eV), the elevation of the energy levels on going from T2-ORH to TF-ORH reduced
the energy offsets (ΔELUMO and ΔEHOMO) between donor and acceptor. The ΔELUMO for the PTB7-Th:TF-ORH blend film was lowered to
0.07 eV, while the ΔELUMO value for the
PTB7-Th:T2-ORH film was slightly larger than that of the PTB7-Th:TF-ORH film
but still smaller than 0.3 eV. Previous studies have indicated that HOMO and
LUMO energy level offsets greater than 0.3 eV are required for efficient
energy transfer between the donor and acceptor.33 However, recent
investigations have shown that effective energy transfer can also occur in
devices with energy offsets below 0.3 eV in which the smaller offset leads to a
small Eloss, allowing the maximum possible open-circuit
voltage (VOC) to be achieved in a given D:A system.34
In our previous study, a small Eloss of ~0.5 eV and thus a
high VOC of 1.05 eV were achieved in a D:A system in
which the DELUMO between donor and
acceptor was smaller than 0.1 eV.18 Table 2 summarized the
physical properties of TF-ORH and PTB7-Th.
Organic
Photovoltaic Properties. The PSCs were fabricated with the inverted device structure: ITO/ZnO
NPs/PEIE/active layer/MoOx/Ag. An active layer was prepared
consisting of PTB7-Th and TF-ORH as donor and acceptor, respectively. PTB7-Th
and TF-ORH were dissolved in chloroform in a ratio of 1:2 at a total solids
concentration of 15 mg mL-1. The current density–voltage (J–V)
and external quantum efficiency (EQE) curves obtained under AM 1.5G
illumination (100 mW cm–2) are shown in Figure 3. The photovoltaic
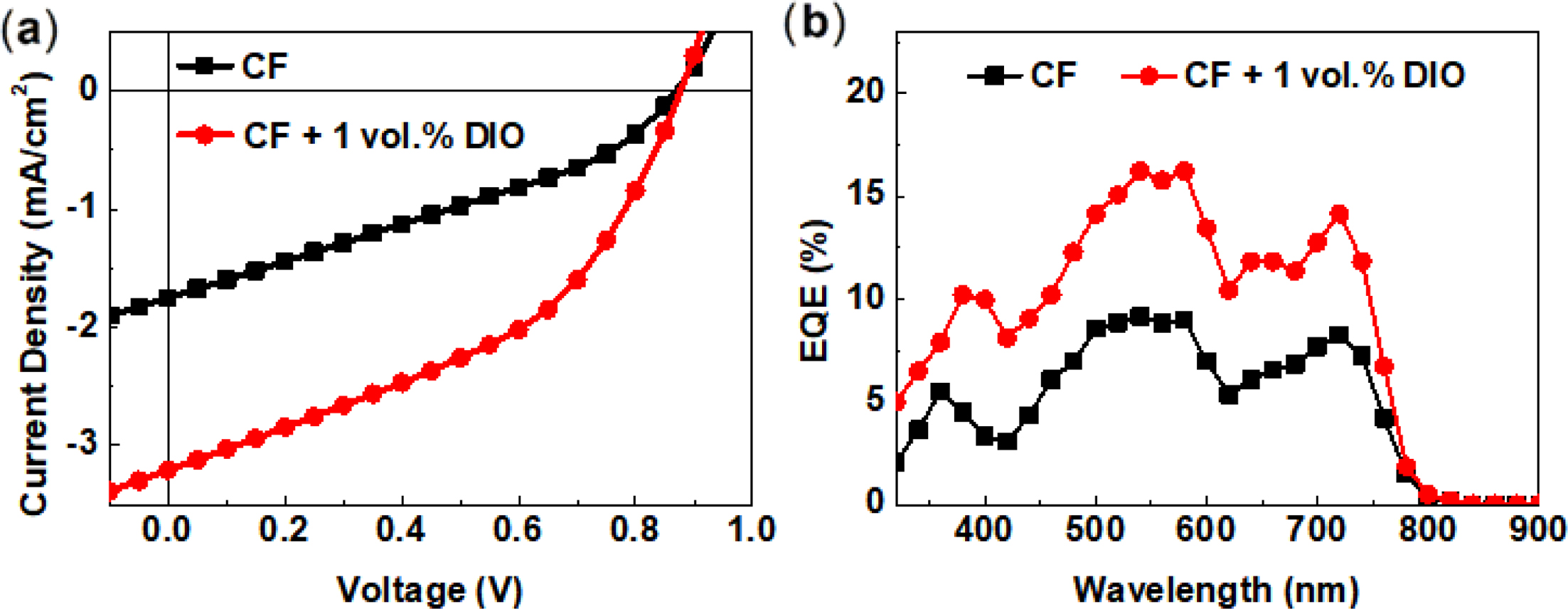
properties of the PTB7-Th:TF-ORH device are summarized in Table 3.
The as-cast device exhibited a PCE of 0.49% with a VOC
of 0.87 V, a short-circuit current (JSC) of 1.75 mA cm–2,
and a fill factor (FF) of 32%. When 1.0 vol% diiodoctane (DIO) was added to the
chloroform solvent as an additive, the device performance was increased upto
1.21% owing to the increased JSC (3.21 mA cm–2)
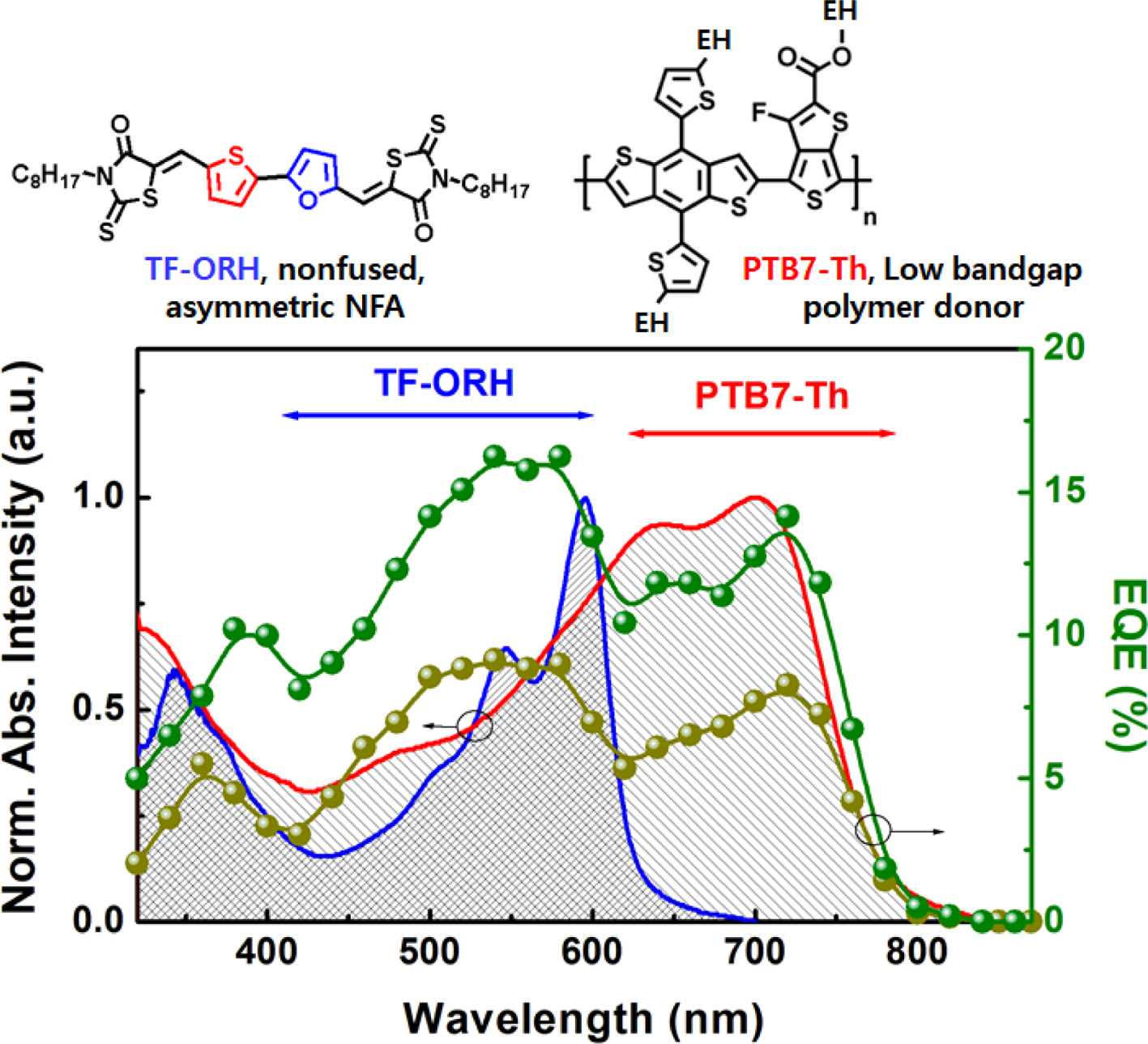
and FF (43%). Moreover, The PTB7-Th:TF-ORH device exhibited a broad EQE profile
from 350–750 nm. The two EQE peaks centered at 540 and 720 nm corresponded to the UV–Vis
absorption maxima of TF-ORH (λmax = 445 nm) and PTB7-Th (λmax = 700 nm), respectively, indicative
of charge generation via both hole and electron transfer.17,30
The VOC of
the PTB7-Th:TF-ORH device was 0.88 V, which was lower than that of the
PTB7-Th:T2-ORH device (1.04 V), but comparable with those reported for
high efficiency devices35 (e.g., PTB7-Th:ITIC (VOC = 0.83
V)).30 The relatively high VOC of the
PTB7-Th:TF-ORH device can be explained by the high-lying LUMO energy level of
TF-ORH and the relatively low Eloss of 0.70 eV, calculated
using the following equation:36 Eloss = Eg,opt
(PTB7-Th)-eVOC. According to the literature,36
few studies have achieved Eloss values below 0.7 eV for
devices prepared using a low bandgap (<1.6 eV) donor polymer and a wide
bandgap (>1.8 eV) NFA.
|
Figure 1 (a) DSC of TF-ORH and T2-ORH; (b) TGA of the TF-ORH. |
|
Figure 2 (a) UV-Vis absorption spectra; (b) CV; (c) energy diagram of the donor and acceptors used in this study |
|
Figure 3 (a) J–V; (b) EQE curves of the PTB7-Th:TF-ORH PSCs. |
|
Table 1 Thermal Properties of TF-ORH |

a
Temperature showing 5% weight loss from the initial weight. b
Temperature at the endothermal melting peak. c
Enthalpic change at the endothermal
melting peak. d
Temperature at the exothermal crystallization peak. c
Enthalpic change at the exothermal crystallization peak. |
|
Table 2 Physical Properties of TF-ORH and PTB7-Th |
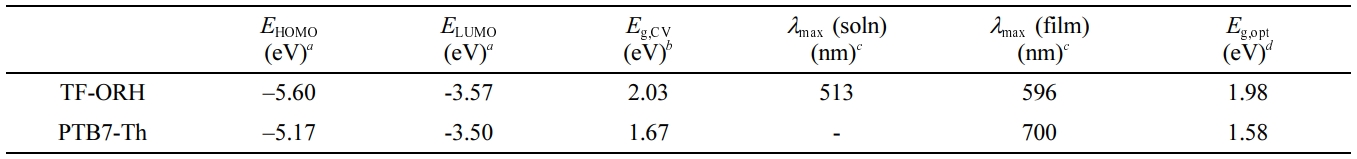
a
EHOMO = –( Eonset,ox – E1/2,Fc + 4.8), ELUMO = –( Eonset,red – E1/2,Fc + 4.8). b
Eg,CV = ELUMO – EHOMO. c
Absorption maxima in chloroform solution and film. d
Eg,opt = 1240/λonset (eV). |
|
Table 3 Photovoltaic Properties of the PSC Devicesa, |
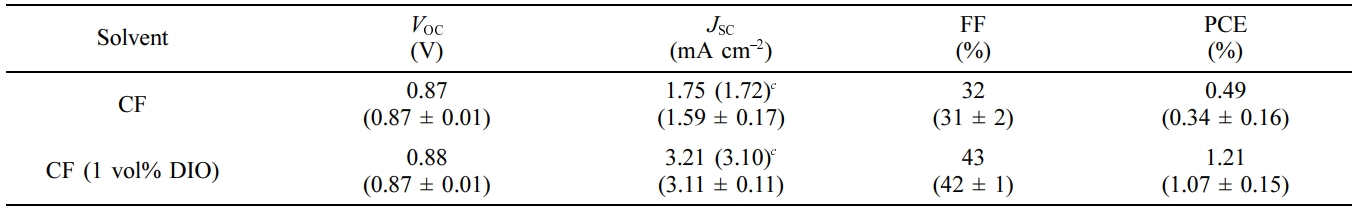
aITO/ZnO
NPs/PEIE/PTB7-Th:TF-ORH/MoOx/Ag. |
We synthesized a nonfused NFA with a simple structure, TF-ORH, containing
a nonfused asymmetrical thiophene-furan core flanked by octyl-substituted
rhodanine end groups. TF-ORH had sufficient solubility and thermal stability
for PSC applications, and exhibited physical properties complementary to those
of the low bandgap polymer donor PTB7-Th. A PSC fabricated using PTB7-Th:TF-ORH
blend film exhibited a PCE of 1.21% with a VOC of 0.88 V and a
small Eloss of 0.70 eV.
- 1. S. Park, H. Ahn, J.-y. Kim, J. B. Park, J. Kim, S. H. Im, and H. J. Son, ACS Energy Lett., 5, 170 (2020).
-
- 2. L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao, K. Xian, H. Y. Woo, Z. Ge, and J. Hou, Adv. Mater., 31, 1903441 (2019).
-
- 3. K. Kranthiraja, H.-Y. Park, K. Gunasekar, W.-T. Park, Y.-Y. Noh, Y.-S. Gal, J. H. Moon, J. Y. Lee, and S.-H. Jin, Macromol. Res., 26, 500 (2018).
-
- 4. S. Nho, D. H. Kim, S. Park, H. N. Tran, B. Lim, and S. Cho, Dyes Pigm., 151, 272 (2018).
-
- 5. H. Sun, T. Liu, J. Yu, T.-K. Lau, G, Zhang, Y. Zhang, M. Su, Y. Tang, R. Ma, B. Liu, J. Liang, K. Feng, X. Lu, X. Guo, F. Gao, and H. Yan, Energy Environ. Sci., 12, 3328 (2019).
-
- 6. Y. Lin and X. Zhan, Mater. Horiz., 1, 470 (2014).
-
- 7. Y.-C. Lin, Y.-J. Lu, C.-S. Tsao, A. Saeki, J.-X. Li, C.-H. Chen, H.-C. Wang, H.-C. Chen, D. Meng, K.-H. Wu, Y. Yang, and K.-H. Wei, J. Mater. Chem. A, 7, 3072 (2019).
-
- 8. J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder, and X. Zhan, J. Am. Chem. Soc., 140, 9140 (2018).
-
- 9. J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li, and Y. Zou, Joule, 3, 1140 (2019).
-
- 10. R. Ma, T. Liu, Z. Luo, Q. Guo, Y. Xiao, Y. Chen, X. Li, S. Luo, X. Lu, M. Zhang, Y. Li, and H. Yan, Sci. China Chem., 63, 325 (2020).
-
- 11. H. Yao, Y. Cui, D. Qian, C. S. Ponseca, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao, and J. Hou, J. Am. Chem. Soc., 141, 7743 (2019).
-
- 12. C. Li, Y. Xie, B. Fan, G. Han, Y. Yi, and Y. Sun, J. Mater. Chem. C, 6, 4873 (2018).
-
- 13. J. Song, C. Li, L. Ye, C. Koh, Y. Cai, D. Wei, H. Y. Woo, and Y. Sun, J. Mater. Chem. A, 6, 18847 (2018).
-
- 14. C. Li, J. Song, L. Ye, C. Koh, K. Weng, H. Fu, Y. Cai, Y. Xie, D. Wei, H. Y. Woo, and Y. Sun, Solar RRL, 3, 1800246 (2019).
-
- 15. X. Li, C. Li, L. Ye, K. Weng, H. Fu, H. S. Ryu, D. Wei, X. Sun, H. Y. Woo, and Y. Sun, J. Mater. Chem. A, 7, 19348 (2019).
-
- 16. C. Li, H. Fu, T. Xia, and Y. Sun, Adv. Energy Mater., 9, 1900999 (2019).
-
- 17. S. Feng, C. E. Zhang, Y. Liu, Z. Bi, Z. Zhang, X. Xu, W. Ma, and Z. Bo, Adv. Mater., 29, 1703527 (2017).
-
- 18. T. Lee, S. Oh, S. Rasool, C. E. Song, D. Kim, S. K. Lee, W. S. Shin, and E. Lim, J. Mater. Chem. A, 8, 10318 (2020).
-
- 19. Y. M. Kim, E. Lim, I.-N. Kang, B.-J. Jung, J. Lee, B. W. Koo, L.-M. Do, and H.-K. Shim, Macromolecules, 39, 4081 (2006).
-
- 20. H. J. Lee, G. E. Park, S. Choi, D. H. Lee, H. A. Um, J. Shin, M. J. Cho, and D. H. Choi, Polymer, 94, 43 (2016).
-
- 21. T. W. Lee, D. H. Lee, J. Shin, M. J. Cho, and D. H. Choi, Polym. Chem., 6, 1777 (2015).
-
- 22. P. Ye, Y. Chen, J. Wu, X. Wu, Y. Xu, Z. Li, S. Hong, M. Sun, A. Peng, and H. Huang, Mater. Chem. Front., 3, 64 (2019).
-
- 23. A. Mahmood, J. Hu, A. Tang, F. Chen, X. Wang, and E. Zhou, Dyes Pigm., 149, 470 (2018).
-
- 24. P. Ye, Y. Chen, J. Wu, X. Wu, S. Yu, W. Xing, Q. Liu, X. Jia, A. Peng, and H. Huang, J. Mater. Chem. C, 5, 12591 (2017).
-
- 25. Suman, A. Bagui, R. Datt, V. Gupta, and S. P. Singh, Chem. Commun., 53, 12790 (2017).
-
- 26. Suman, A. Bagui, A. Garg, B. Tyagi, V. Gupta, and S. P. Singh, Chem. Commun., 54, 4001 (2018).
-
- 27. Y. Eom, C. E. Song, W. S. Shin, S. K. Lee, and E. Lim, J. Ind. Eng. Chem., 45, 338 (2017).
-
- 28. C. Li, T. Xia, J. Song, H. Fu, H. S. Ryu, K. Weng, L. Ye, H. Y. Woo, and Y. Sun, J. Mater. Chem. A, 7, 1435 (2019).
-
- 29. C. Li, J. Song, Y. Cai, G. Han, W. Zheng, Y. Yi, H. S. Ryu, H. Y. Woo, and Y. Sun, J. Energy Chem., 40, 144 (2020).
-
- 30. T. Lee, Y. Eom, C. E. Song, I. H. Jung, D. Kim, S. K. Lee, W. S. Shin, and E. Lim, Adv. Energy Mater., 9, 1804021 (2019).
-
- 31. O. Kwon, J. Jo, B. Walker, G. C. Bazan, and J. H. Seo, J. Mater. Chem. A, 1, 7118 (2013).
-
- 32. D. Liu, B. Kan, X. Ke, N. Zheng, Z. Xie, D. Lu, and Y. Liu, Adv. Energy Mater., 8, 1801618 (2018).
-
- 33. M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger, and C. J. Brabec, Adv. Mater., 18, 789 (2006).
-
- 34. T. Zhang, X. Zhao, D. Yang, Y. Tian, and X. Yang, Adv. Energy Mater., 8, 1701691 (2018).
-
- 35. Suman and S. P. Singh, J. Mater. Chem. A, 7, 22701 (2019).
-
- 36. J. Zhang, L. Zhu, and Z. Wei, Small Methods, 1, 1700258 (2017).
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2020; 44(5): 741-746
Published online Sep 25, 2020
- 10.7317/pk.2020.44.5.741
- Received on Jun 16, 2020
- Revised on Jul 16, 2020
- Accepted on Jul 22, 2020
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
- Supporting Information
Introduction
Experimental
Results and Discussion
- References
Shared
Correspondence to
- Eunhee Lim
-
Department of Chemistry, Kyonggi University, 154-42 Gwanggyosan-ro, Yeongtong-gu, Suwon 16227, Korea
- E-mail: ehlim@kyonggi.ac.kr
- ORCID:
0000-0002-2321-7072
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr