






- Synthesis of Bio-based Poly(ethylene 2,5-furandicarboxylate) in a Kneader Reactor and its Melt Spinning
Seongil Yu, Jae-Chul Lee, Seungjae Ahn, and Ki-Young Kim†

Human Convergence Technology R&D Department, Korea Institute of Industrial Technology (KITECH), 143, Hanggaul-ro, Sangnok-gu, Ansan-si, Gyeonggi-do 15588, Korea
- 바이오 기반의 Poly(ethylene 2,5-furandicarboxylate)의 Kneader 반응기에서 합성과 용융방사
유성일 · 이재철 · 안승재 · 김기영†
한국생산기술연구원 휴먼융합연구부문
There is a growing interest in
using bio-based raw materials as a replacement for petrochemical materials.
Bio-based materials produced from plant resources are considered
environmentally sustainable resources. Poly(ethylene 2,5-furandicarboxylate)
(PEF) is now widely used and is considered the main substitute for
poly(ethylene terephthalate) (PET). In this study, PEF was polymerized in a
flask and a kneader reactor, and the properties of PEF were characterized. In
the kneader reactor, PEF was synthesized effectively, and the resultant
melt-spun fibers with a tensile strength of 1.80 g/den and a strain of
114.25% were obtained, confirming the possibility of utilizing PEF as a fiber.
석유화학물질을 사용하는 것에서 벗어나기 위하여 바이오 기반의
원료를 사용하는 것에 대한 관심이 높아지고 있다. 식물자원으로부터 생산되는 바이오 기반의 원료는 친환경적이며
지속 가능한 자원으로 여겨진다. 현재 널리 사용되고 있는
poly(ethylene terephthalate)(PET)의 대체 후보로 poly(ethylene
2,5-furandicarboxylate)(PEF)가 기대되고 있다. 본 연구는, 플라스크와 kneader 반응기에서 PEF를 중합하여 특성을 비교, 확인하였다. Kneader 반응기에서 효과적으로 PEF를 합성할 수 있었으며, 결과물을 용융 방사한 섬유는 1.80 g/den의 인장강도와, 114.25%의 변형률을 보여주어 PEF를 섬유로 사용할 수 있는
가능성을 확인하였다.
Poly(ethylene 2,5-furandicarboxylate) (PEF) was
polymerized in a flask and a kneader reactor, and the properties of PEF were
characterized. In the kneader reactor, PEF was synthesized effectively, and the
resultant melt-spun fibers with a tensile strength of 1.80 g/den and a strain of 114.25% were
obtained, confirming the possibility of utilizing PEF as a fiber.
Keywords: bio-based polymers, poly(ethylene 2,5-furandicarboxylate), 2,5-furandicarboxylic acid, polyesters, kneader reactor.
This work was supported by the Technology Innovation
Program (10077966, Development of Biomass-based Polyethylenefuranoate (PEF)
fiber) funded by the Ministry of Trade Industry & Energy (MOTIE, Korea).
In recent years, there has been growing interest in sustainable bio-based
materials as a viable alternative to fossil fuels with limited reserves.
Biomass-based materials, such as corn, sugar cane, woody plant resources, palm,
seaweed, and other plant resources are considered an important resource for
replacing petroleum resources because of their environmental evolution and
sustainability.1 These biomass-based materials can be synthesized
with lactic acid, isosorbide, succinic acid, dodecanedioic acid, and ethylene
glycol, which can then be used as building blocks for bio-based polymers such
as poly(lactic acid) (PLA), poly(hydroxy alkanoates) (PHA), and poly(butylene
succinate) (PBS).1-3
In modern society, poly(ethylene terephthalate) (PET) is used in a wide
variety of fields, ranging from beverage bottles, films, and textiles.
Replacing PET with bio-based materials can play a major role in environmentally
sustainable development. The most promising alternative to PET is poly(ethylene
2,5-furandicarboxylate) (PEF).4,5 PEF is produced by
polycondensation from ethylene glycol (EG) and 2,5-furandicarboxylate (FDCA).
EG and FDCA can be obtained from renewable bio-based materials and subsequently
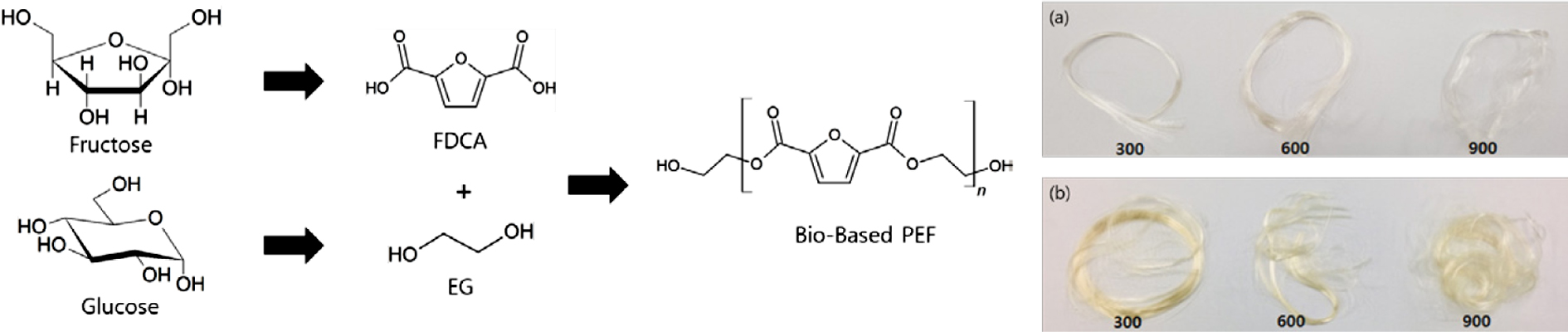
used to make 100% bio-based polymers (Figure 1).5-7 FDCA is
synthesized from fructose, the plant material, via 5-hydroxymethylfufural
(HMF).7,8 In addition, it can be used as an alternative to
terephthalic acid (TPA) due to its bio-based aromatic ring (furan ring).9,10
While
PEF has a similar structure to PET, it has better gas barrier properties for
use in the packaging and film market11,12 compared to PET with high
thermal properties. Moreover, the PEF fiber is a promising sustainable
bio-based material because it can be spun and dyed using conventional PET
manufacturing facilities for apparel and industrial non-woven fabrics.
In this study, PEF was synthesized and spun using laboratory mixing
extruder to simulate the spinning process for PEF fibers, and the PEF fibers
properties were characterized. To fabricate the fibers, a PEF polymer with a
higher molecular weight, narrow molecular weight distribution, high purity, and
higher elongation properties is required. Polymerization was carried out using
a kneader reactor (Figure 2). Since the twin shaft of the kneader reactor was
designed to effectively mix high-viscosity reactants (Figure 2(right)), it is
expected that the byproduct can be removed smoothly, and the molecular weight
of the polymers can be efficiently increased by the kneading process. In
addition, PEF was synthesized in a flask as comparison to the kneader reactor
method, and the resultant fiber was fabricated by spinning at various winding
speeds. By analyzing and comparing the characteristics of fibers, it has been
confirmed that PEF can be applied to textile systems.

|
Figure 1 Bio-based PEF from bio-based EG and bio-based FDCA. |

|
Figure 2 Kneader reactor (left) and twin shaft of kneader reactor
(right). |
Materials. 2,5-furandicarboxylate (FDCA, sheobiotech), ethylene glycol (EG, extra
pure grade, Ducksan Co., Ltd.), titanium (IV) isopropoxide (TIS, 99.999%,
Sigma-Aldrich Co.), and antimony (III) oxide (99.83%, Sooyangchemtec Co., Ltd.)
were used as received.
Synthesis
of PEF. PEF synthesis was
conducted in two steps: After the esterification reaction in the nitrogen purge
state, PEF was synthesized by a polycondensation reaction in vacuum (Figure 3).
Synthesis
of PEF in a Flask (PEF-F): 1 mol of FDCA
(156.09 g), 2 mol of EG (124.14 g), 0.06 g (400 ppm of FDCA weight) of TIS, and
0.78 g (0.5% of FDCA weight) of SONGNOX 1680 (heat stabilizer) were added to a
500 mL three-necked round bottom flask and mixed well. The mixture was stirred
at 200 rpm in a nitrogen atmosphere for 10 min. With the temperature raised to
180 °C, the reaction was carried out for 1 h. When the reaction started,
the water produced as a byproduct was removed through the condenser. Next, the
reaction was first carried out at 190 °C for 1 h followed by 200 °C
for 2 h, and the reaction product gradually became a brown transparent
liquid. Thereafter, the temperature was increased to 230 °C, and the
vacuum was gradually set to maintain a pressure of <5 mbar for
polycondensation. The reaction was allowed to proceed at 230 °C for 1 h,
240 °C for 1 h, and 250 °C for 2 h. The EG that was produced as
byproduct was removed using a condenser. After the reaction completed, the PEF
was quenched and no purification was performed.
Synthesis
of PEF in the Kneader Reactor (PEF-K): A kneader reactor
was used with a CRP 2.5 batch from LIST. 13 mol of FDCA (2029.17 g), 32.5
mol of EG (2017.28 g), 0.81 g of TIS and 10.15 g of SONGNOX 1680 were added and
mixed by rotating the shaft while purging with nitrogen. While the molar ratio
of FDCA to EG was 1:2 when reacted in a flask, the ratio was 1:2.5 when reacted
in a kneader reactor as it took a long time to heat due to the large volume and
to compensate for the evaporation of EG. The reaction in a flask was carried
out at 180 °C for 1 h, 190 °C for 1 h, and 200 °C for 2 h. As the reaction
progressed, water escaped out and the reactant turned into a clear, brown
liquid. It was then heated to 230 °C and vacuumed to <5 mbar. The reaction
was carried out in a vacuum at 230 °C for 1 h, 240 °C for 1 h, and
250 °C for 2 h. After the reaction completed, the screw was rotated while
cooling to pulverize the PEF. No purification process was performed.
Melt
Spinning of PEF. PEF was melt spun
using a laboratory mixing extruder (Dynisco). A 1/8 inch spinneret was used and
spun at a rotor temperature of 210 °C and a header temperature of 230 °C.
The distance between the spinneret and winder was 40 cm, and the winder speed
was 300, 600, and 900 m/min respectively.
Characterization. Gel permeation
chromatography (GPC) was performed using an EcoSEC HLC-8320 (Tosoh),
RI-detector, and TSKgel Super AWM-H column. Calibration was performed using
poly(methyl methacrylate) (659000, 121600, 30620, 6820, 4900, 2680, 850, and
162 g/mol) standards with a narrow molecular weight distribution.
Hexafluoroisopropanol (HFIP) containing 0.01 N sodium trifluoroacetate was used
as a solvent with the mobile phase of a flow fate of 0.3 mL/min at 40 ℃.
All polymer samples were dissolved in the solvent (6 mg/mL) and filtered
through a 0.45 μm PTFE membrane before injection.
Fourier-transform infrared (FTIR) spectroscopy was performed 32 times in
the range of 4000-650 cm-1 using the Thermo Nicolet iS50 ATR
method. Intrinsic viscosity (IV) measurements were performed using a Ubbelohde
viscometer at 25 °C. PEF samples (5, 2.5, 1.25 g/dL) were dissolved in a
phenol/1,1,2,2-tetrachloroethane (3/2, w/w) mixture at 90 °C. IV was
calculated by obtaining the reduced viscosity (ηred), inherent
viscosity (ηinh) and using the
method of obtaining the y-intercept of two viscosities.

where c is the concentration of the solution, t is the flow
time of the solution, and t0 is the flow time of the pure
solvent. 1H nuclear magnetic resonance (1H NMR, Varian
VXRUnity NMR 400 MHz) was measured by dissolving the sample in DMSO-d6.
Differential scanning calorimetry (DSC) was performed using a TA Instruments
Q100 and an aluminum standard pan. Scans were conducted with nitrogen at a
heating rate of 10 °C/min in the temperature ranging from 30 to
300 °C. Thermogravimetric analysis (TGA) was performed using a TA
Instruments Q500 and platinum pan. The samples were heated to 800 °C at a
rate of 20 °C/min with nitrogen. The spun fibers were characterized using
a testing system for a single fiber (FAVIMAT). The linear density was tested
with a pretension of 0.5 cN/tex and a speed of 2 mm/min. The tensile
strength was tested at a pretension of 0.5 cN/tex and a speed of 20 mm/min.

|
Figure 3 Esterification and polycondensation reactions of PEF. |
The structures of PEF synthesized in a kneader reactor and flask were
analyzed. The functional groups of PEF were identified through FTIR, and the
absorption peaks corresponding to the respective functional groups were
measured (Figure 4(a)). The stretching band of the furan ring =C-H was 3123 cm-1,
the bending band of C=C was at 968, 768 cm-1, and the stretching
band was at 1578 cm-1. The stretching bands of C=O and C-O of the
ester groups were 1733 and 1262 cm-1, respectively. These results
are consistent with the results of other studies.13,14 PEF-F and
PEF-K did not show any difference in FTIR spectra, and they were synthesized
normally in the flask and kneader reactor. The structure of PEF was confirmed
by 1H NMR (Figure 4(b)). The peak at 7.4 ppm (a) was attributed to
the furan ring and the peak at 4.6 ppm (b) was attributed to the -OCH2CH2O-
group.9,15 Small peaks at 4.4 and 3.7 ppm were observed by the end
groups, and peak at 1.2 ppm was attributed to catalysts and additives, mainly
to the thermal stabilizer. The thermal stabilizer was not removed for the
subsequent spinning process to prevent thermal degradation of the polymers.
Based on the GPC analysis, the average molecular weight (Mn)
of PEF-F and PEF-K were 18177 and 22376 g/mol, respectively (Table 1).
Although the scale of the kneader reactor was larger, the Mn
of PEF-K was higher than that of PEF-F, suggesting that the kneader reactor was
effectively mixed and kneaded.
The glass transition temperature (Tg) and melting temperature
(Tm) of PEF were measured by DSC (Figure 5(a)). The Tg
of PEF-F was observed at 84 °C. The Tg of PEF-K was not
evident in the first heat curve, but was observable in the second heat curve at
88 °C. In the first heat curve, Tm of PEF-F and PEF-K were
211 and 213 °C, respectively. In addition, since PEF-F was quenched after
synthesis, it can be seen that a peak due to crystallization peak appears
before Tm peak. In the second heat curve, the Tm
values of PEF-F and PEF-K were very small. This is due to the fact that the
crystallization has hardly occurred due to the high speed scanning. The
difference between the first heat curve and the second heat curve is due to the
different thermal histories. The properties of the polymer appear in the curve
of the second scan with the thermal history removed, however the information
from the first curve is also important because the spinning process requires
information on the synthesized PEF properties. The thermal decomposition
temperatures of PEF-F and PEF-K in the TGA thermograms were both 408 °C,
having started at approximately 350 °C (Figure 5(b)). PEF-F and PEF-K
showed similar thermal decomposition characteristics. Thermal decomposition
behavior is most affected by the structure of the main chain, so the effect due
to the molecular weight is minimal.
Based on the thermal properties of PEF-F and PEF-K, the spinning
temperature was 230 °C, approximately 20 °C higher than Tm
of PEF. Figure 6 shows pictures of melt-spun PEF-F and PEF-K fibers at various
winding speeds. The spun fibers were measured for their denier and tensile
properties using a single fiber tensile tester. The results are shown in Table
2. As the winding speed increased, the tensile strength increased (Figure 7(b))
while the denier and strain decreased (Figure 7(a), (c)). The PEF-F showed an
elongation to fiber fracture of approximately 2%, and was deemed to be very
brittle. It could not be used because of the impossibility of the drawing
process thereafter. PEF-K however, showed a higher failure strain of
approximately 240-110%. This means that drawing processing is subsequently
possible for further improvement of the fiber tensile properties. PEF-K has
significantly higher tensile strength than PEF-F. Due to the molecular
structure of the main chain, PEF is more brittle than PET.11 The
brittle nature of PEF results in PEF-F with little elongation and also presents
difficulties in the drawing process with fiber breakage. If the increased
molecular weight of PEF-K (as shown in Table 1) is sufficient to entangle the
molecular chains, elongation can be increased. In polymers, higher elongations,
such as PEF-K, imply that the polymer chain has sufficient molecular weight and
entanglement to withstand the applied loads.16 The tensile
stress-strain curves illustrates the brittle nature of PEF-F fibers (Figure
8(a)), whereas the PEF-K showed ductile fracture characteristics. With a large
elongation to fracture in PEF-K due to the molecular orientation and strain
hardening, this leads to increased tensile strength (Figure 8(b)).

|
Figure 4 (a) FTIR spectra; (b) 1
H NMR spectrum of PEF-F and
PEF-K. |

|
Figure 5 (a) DSC heat curves; (b) TGA thermograms of PEF-F and
PEF-K. |

|
Figure 6 Melt spun PEF fibers at winding speeds of 300, 600, and
900 m/min: (a) PEF-F; (b) PEF-K. |

|
Figure 7 Tensile properties of PEF-F and PEF-K fibers: (a) denier;
(b) tensile strength; (c) strain as winding speed. |

|
Figure 8 Stress-strain curves of (a) PEF-F; (b) PEF-K fibers. |


PEF, a bio-based polymer, was synthesized in both a flask and a kneader
reactor. Although each PEF showed no structural difference, the PEF synthesized
in the kneader reactor showed higher molecular weight compared to the flask.
This seems to be due to the effective mixing and kneading of the reactants in
the kneader reactor. The synthesized PEF fibers were fabricated by melt
spinning. As the winding speed increased, the fiber fineness tended to
decrease. PEF-K showed higher tensile strength and strain than PEF-F. The PEF-K
fiber at a winding speed of 300 m/min was found to have an elongation of more
than 200% due to the high molecular weight and entanglement of the polymer.
This study shows the potential and viability of PEF polymers as fibrous
materials. Further research on bio-based PEF polymers is expected to contribute
to the textile industry in the foreseeable future.
- 1. T. Iwata, Angew. Chem. Int. Ed. Engl., 54, 3210 (2015).
-
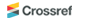
- 2. E. T. Vink, K. R Rabago, D. A. Glassner, and P. R. Gruber, Polym. Degrad. Stab., 80, 403 (2003).
-
- 3. R. W. Lenz and R. H. Marcjessault, Biomacromolecules, 6, 1 (2005).
-
- 4. A. Gandini, A. J. Silvestre, C. P. Neto, A. F. Sousa, and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 47, 295 (2009).
-
- 5. A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho, and A. J. D. Silvestre, Polym. Chem., 6, 5961 (2015).
-
- 6. N. Li, Y. Zheng, L. Wei, H. Teng, and J. Zhou, Green Chem., 19, 682 (2017).
-
- 7. C. Triebl, V. Nikolakis, and M. Ierapetriou, Comput. Chem. Eng., 52, 26 (2013).
-
- 8. M. J. Climent, A. Corma, and S. Iborra, Green Chem., 13, 520 (2011).
-
- 9. A. F. Sousa, M. Matos, C. S. Freire, A. J. Silvestre, and J. F. Coelho, Polymer, 54, 513 (2013).
-
- 10. E. Gubbels, L. Jasinska-Walc, and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 51, 890 (2013).
-
- 11. S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel, and W. J. Koros, Macromolecules, 47, 1383 (2014).
-
- 12. H. Nakajima, P. Dikstra, and K. Loos, Polymers, 9, 523 (2017).
-
- 13. M. Gomes, A. Gandini, A. J. Silvestre, and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 49, 3759 (2011).
-
- 14. M. Matos, A. F. Sousa, A. C. Fonseca, C. S. R. Freire, J. F. J. Coelho, and A. J. D. Silvestre, Macromol. Chem. Phys., 215, 2175 (2014).
-
- 15. Z. Jia, J. Wang, L. Sun, J. Zhu, and X. Liu, J. Appl. Polym. Sci., 135, 46076 (2018).
-
- 16. R. W. Nunes, J. R. Martin, and J. F. Johnson, Polym. Eng. Sci., 22, 205 (1982).
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2020; 44(5): 695-700
Published online Sep 25, 2020
- 10.7317/pk.2020.44.5.695
- Received on May 14, 2020
- Revised on Jun 29, 2020
- Accepted on Jun 30, 2020
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Ki-Young Kim
-
Human Convergence Technology R&D Department, Korea Institute of Industrial Technology (KITECH), 143, Hanggaul-ro, Sangnok-gu, Ansan-si, Gyeonggi-do 15588, Korea
- E-mail: kkim@kitech.re.kr
- ORCID:
0000-0002-9255-3264
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr