






- Influence of CN Substitution on DPP-furan-based Small-molecule Acceptors for Polymer Solar Cells
Eunhee Lim†

Department of Chemistry, Kyonggi University, 154-42 Gwanggyosan-ro, Yeongtong-gu, Suwon 16227, Korea
- 고분자 태양전지용 DPP와 Furan 기반 단분자 어셉터의 CN 치환기 효과
임은희†
경기대학교 화학과
In this study, two
small-molecules, p- and o-DPP-F-PhCN, were synthesized via a
Suzuki coupling reaction and used as nonfullerene acceptors (NFAs) for
poly(3-hexylthiophene) (P3HT)-based polymer solar cells (PSCs). The physical
properties of the molecules were examined in terms of the substituent position
and the effect of the furan moiety. The introduction of a furan moiety resulted
in higher phase-transition temperatures and higher-lying molecular orbital
energy levels of the molecules. Substitution at the ortho position also
elevated the energy levels of the molecules, resulting in the highest-lying
values for o-DPP-F-PhCN. A relatively enhanced and red-shifted UV–vis
absorption of o-DPP-F-PhCN indicated its stronger molecular aggregation.
PSCs based on two DPP-F-PhCNs exhibited similar device efficiencies. The
stronger aggregation behavior of o-DPP-F-PhCN led to a device with a
better external quantum efficiency profile; however, the enhanced orbital
interactions and resulting stabilized the lowest unoccupied molecular orbital
level of o-DPP-F-PhCN led to a relatively low open-circuit voltage.
본 연구에서는, 스즈키
짝지음 반응을 이용하여 p- 및 o-DPP-F-PhCN의 두 단분자를 합성하여 고분자 태양전지의
비풀러렌계 어셉터로 사용하였다. 치환기의 위치 및 퓨란 효과에 따른 단분자의 물리적 특성 변화를 살펴보았다. 퓨란 그룹의 도입으로 단분자는 더 높은 상전이 온도와 HOMO 및 LUMO 준위를 나타내었다. o-치환 역시 분자의 에너지
준위를 높게 함으로써, o-DPP-F-PhCN의 에너지 준위가 가장 높게 나타났다. 상대적으로 강하고 장파장 이동된 UV-vis 흡수는 o-DPP-F-PhCN의 강한 분자 응집을 나타낸다. P3HT와 DPP-F-PhCN들을 각각 고분자 도너와 비풀러렌계 어셉터로 사용한 고분자 태양전지를 제작하였고, 두 DPP-F-PhCN이 유사한 효율을 나타내었다. o-DPP-F-PhCN의 강한 응집 거동은 외부양자효율에 유리하였으나, 향상된
오비탈 상호작용에 따른 안정화된 LUMO 준위로 인해 상대적으로 낮은 개방전압 값을 나타내었다.
Two DPP-based small molecules of p- and o-DPP-F-PhCNs
were synthesized via a Suzuki coupling reactions and showed similar device
efficiencies when used as nonfullerene acceptors for
poly(3-hexylthiophene)-based polymer solar cells.
Keywords: polymer solar cells, organic photovoltaic cells
This work was supported by Kyonggi University Research
Grant 2017.
Information is available regarding 1H and 13C NMR
spectra, UV-vis absorption, and
all-OSC characterization. The materials are available via the Internet at
http://journal.polymer-korea.or.kr.
PK_2020_044_03_408_Supporting_Information.pdf (612 kb)
Supplementary Information
Recently, nonfullerene acceptors (NFAs) have been spotlighted as
replacements for the traditional fullerene acceptors in polymer solar cells
(PSCs). Most successful examples are acceptor–donor–acceptor (A–D–A)-type
small-molecule NFAs composed of ladder-type electron-donating cores (e.g.,
indacenodithiophene or indacenodithieno[3,2-b]thiophene) and
electron-withdrawing dye end groups (e.g., rhodanine (RH) or
1,1-dicyanomethylene-3-indanone).1,2 Continuous and intensive
efforts toward the development of various A–D–A-type NFAs has opened a path to
the development of high-efficiency PSCs. A certified power conversion efficiency (PCE)
of 17.29% was achieved in 2018 using a solution-processed tandem PSC in which
two NFAs (F-M and COi8DFIC) were used in the rear (PBDB-T:F-M) and
front (PTB7-Th:COi8DFIC:PC71BM) subcells,
respectively, resulting in high external quantum efficiency (EQE) responses
greater than 70% in the whole UV–vis and infrared absorption ranges covering
wavelengths as long as 1000 nm.3 More recently, a high PCE of 17.0%
was also demonstrated in single-junction PSCs composed of PBDBTF and BTP-4Cl-12
as a polymer donor and an NFA, respectively, through side-chain engineering.4
However, most of the reported high-efficiency NFAs have been composed of
ladder-type aromatic rings with extended p-conjugation, which
requires multiple synthesis steps and inevitably involves high synthesis costs;
thus, this approach is not applicable to the future commercialization of PSCs.
Consequently, NFAs based on nonfused core systems have recently been attracting
increasing interest.1,5-8
Side-chain engineering (i.e., length and bulkiness) and/or substituent
effects (i.e., electron-donating or -withdrawing groups) have been explored to
fine-tune the physical properties of organic semiconducting materials,
including their solubility and molecular aggregation behavior.1,4,9
The position of side chains and substituents can also affect the physical
properties of the resultant molecules. For example, Janssen et al.
systematically investigated the influence of the position of the alkyl side
chain on the photovoltaic properties of 2,5-diketopyrrolo[3,4-c]pyrrole
(DPP)-based small molecules10 and found that the morphology and
crystallization of the small molecules varied according to the side-chain
position. In addition, our group has reported the effect of hexyl or cyanide
(CN) groups on the photovoltaic properties of benzothiadiazole–thiophene-based
small molecules (2-, 3-, and 4-hexyl-substituted DH5TBs)11 or
DPP–thiophene-based small molecules (p-, m-, and o-DPP-PhCNs),
respectively.12
In the present work, we developed two small molecules, p- and o-DPP-F-PhCN,
in which electron-withdrawing CN groups were introduced at different positions
(para and ortho, respectively) on the phenyl end groups, and observed their
different molecular packing behaviors and photovoltaic properties. In
particular, we used differential scanning calorimetry (DSC), UV–vis absorption
spectroscopy, and cyclic voltammetry (CV) measurements to systematically
investigate how the introduction of a furan moiety altered the physical
properties of the molecules.
Materials. Tris(dibenzylideneacetone)dipalladium(0)
(Pd2 (dba)3), 4-cyanophenylboronic acid pinacol ester,
tri-tert-butylphosphonium tetrafluoroborate (P(t-Bu)3 × HBF4),
and potassium phosphate tribasic (K3PO4) were purchased
from Sigma Aldrich. 2-Cyanophenylboronic acid pinacol ester was purchased from
Alfa Aesar. All chemicals were used without further purification, and all
reactions were performed under a nitrogen atmosphere with anhydrous solvents.
Synthesis. DPP-F-Br was synthesized
according to the literature.13,14 p- and o-DPP-F-PhCN
were synthesized using a palladium-catalyzed Suzuki coupling reaction between
DPP-F-Br and boronic esters, 4- and 2-cyanophenylboronic acid pinacol ester,
respectively.12,15
Synthesis
of p-DPP-F-PhCN: A degassed aqueous
solution (1.3 mL) of K3PO4 (0.40 g, 1.9 mmol) was
added to 12 mL of degassed tetrahydrofuran (THF) solution of DPP-F-Br
(0.40 g, 0.62 mmol), Pd2(dba)3 (0.016 g, 0.017 mmol), P(t-Bu)3 × HBF4
(0.010 g, 0.033 mmol), and 4-cyanophenylboronic acid pinacol ester
(0.45 g, 2.5 mmol) under N2 atmosphere. After the mixture
was heated to 80 oC for 12 h, the reaction was cooled to room
temperature. The mixture was extracted with dichloromethane and water. The
collected organic layer was dried over MgSO4. After removing the
solvent under reduced pressure, the crude product was purified by column
chromatography on silica using dichloromethane as the eluent, to afford p-DPP-F-PhCN
as a dark green powder (0.37 g, yield 86%). 1H NMR (CDCl3,
400 MHz): δ (ppm) 8.43 (d, 2H), 7.85 (d, 4H), 7.74 (d,
4H), 7.12 (d, 2H), 4.16 (m, 4H), 1.89 (m, 2H), 1.23-1.46 (m,
16H), 0.90 (t, 6H), 0.83 (t, 6H). 13C NMR (CDCl3,
100MHz): δ (ppm) 161.03, 154.58, 145.26, 133.16, 133.06, 132.85, 124.71,
122.46, 118.50, 111.88, 111.83, 108.10, 46.70, 39.48, 30.37, 28.54, 23.59,
23.16, 14.03, 10.56. Anal. calcd for C44H46N4O4:
C, 76.05; H, 6.67; N, 8.06; O, 9.21. Found: C, 76.22; H, 6.72; N, 8.10.
Synthesis
of o-DPP-F-PhCN: o-DPP-F-PhCN was prepared in the same manner as p-DPP-F-PhCN
using a degassed aqueous solution (1.1 mL) of K3PO4 (0.33
g, 1.6 mmol), 9.4 mL of degassed THF solution of DPP-F-Br (0.33 g, 0.51 mmol),
Pd2(dba)3 (0.013 g, 0.014 mmol), P(t-Bu)3
× HBF4 (8.0 mg, 0.027 mmol), and 2-cyanophenylboronic acid pinacol
ester (0.47 g, 2.04 mmol), to afford o-DPP-F-PhCN as a purple powder
(0.31 g, yield 89%). 1H NMR (CDCl3, 400 MHz): δ
(ppm) 8.35 (d, 2H), 7.94 (d, 2H), 7.72 (dd, 2H), 7.64 (td,
2H), 7.53 (d, 2H), 7.39 (td, 2H), 4.11 (m, 4H), 1.80 (m,
2H), 1.13-1.36 (m, 16H), 0.81 (t, 6H), 0.73 (t, 6H). 13C
NMR (CDCl3, 100 MHz): δ (ppm) 160.95, 152.28,
144.81, 134.55, 133.16, 131.72, 128.54, 126.53, 122.22, 118.55, 113.94, 108.15,
107.83, 46.48, 39.51, 30.34, 28.54, 23.57, 23.15, 14.02, 10.58. Anal. calcd for
C44H46N4O4: C, 76.05; H, 6.67; N,
8.06; O, 9.21. Found: C, 76.56; H, 6.74; N, 8.22.
Physical
Measurements. NMR spectra were recorded on a Bruker AVANCE II 400
spectrometer. Elemental analyses were performed with a Flash EA 1112 series
from Thermo Electron Corporation. Thermogravimetric analysis (TGA) and DSC were
performed on a TGA/DSC 1 thermogravimetric analyzer from Mettler-Toledo Inc.
under a nitrogen atmosphere at a heating or cooling rate of 10 °C/min.
UV-vis spectra were obtained using a Shimadzu UV-2550 spectrometer. The
electrochemical properties of the small molecules were studied by CV with a BAS
100B electrochemical analyzer. The films were prepared by dip-coating the small
molecule solution onto a Pt working electrode. Detailed experimental methods
are given in previous reports.12
Fabrication
of PSCs. In this study, we fabricated the devices with the
structures of ITO/PEDOT:PSS/poly(3-hexylthiophene) (P3HT):acceptor/LiF/Al. The
ITO-coated glass substrates were pre-treated in a UV-ozone oven for 15 min. A
layer of PEDOT:PSS (~30 nm) was spin-coated on top of the ITO-coated glass
substrates. The active layer was spin-cast at 3000 rpm from a chloroform
solution of donor and acceptor with a total solids concentration of 15 mg mL-1.
The average thickness of the active layers (~100 nm) was measured with an
Alpha-Step IQ surface profiler. A LiF (~0.5 nm) and Al (~100 nm)
layer were directly deposited on the active layer under a vacuum of ~10-6
Torr. The effective area of all devices was measured to be 9 mm2.
The current-density vs. voltage (J–V) and EQE curves were
recorded using the same methods described in our previous report.12
Synthesis
and Thermal Properties. The two small molecules p-
and o-DPP-F-PhCN were synthesized via a Suzuki coupling reaction between
the dibromides of a DPP-furan-based core (DPP-F-Br) and two corresponding
boronic esters (4- and 2-cyanophenylboronic acid pinacol ester, respectively)
using a Pd catalyst (Scheme 1). The synthesized small molecules were
successfully characterized by 1H- and 13C NMR
spectroscopy (Figures S1–S2) and elemental analysis.
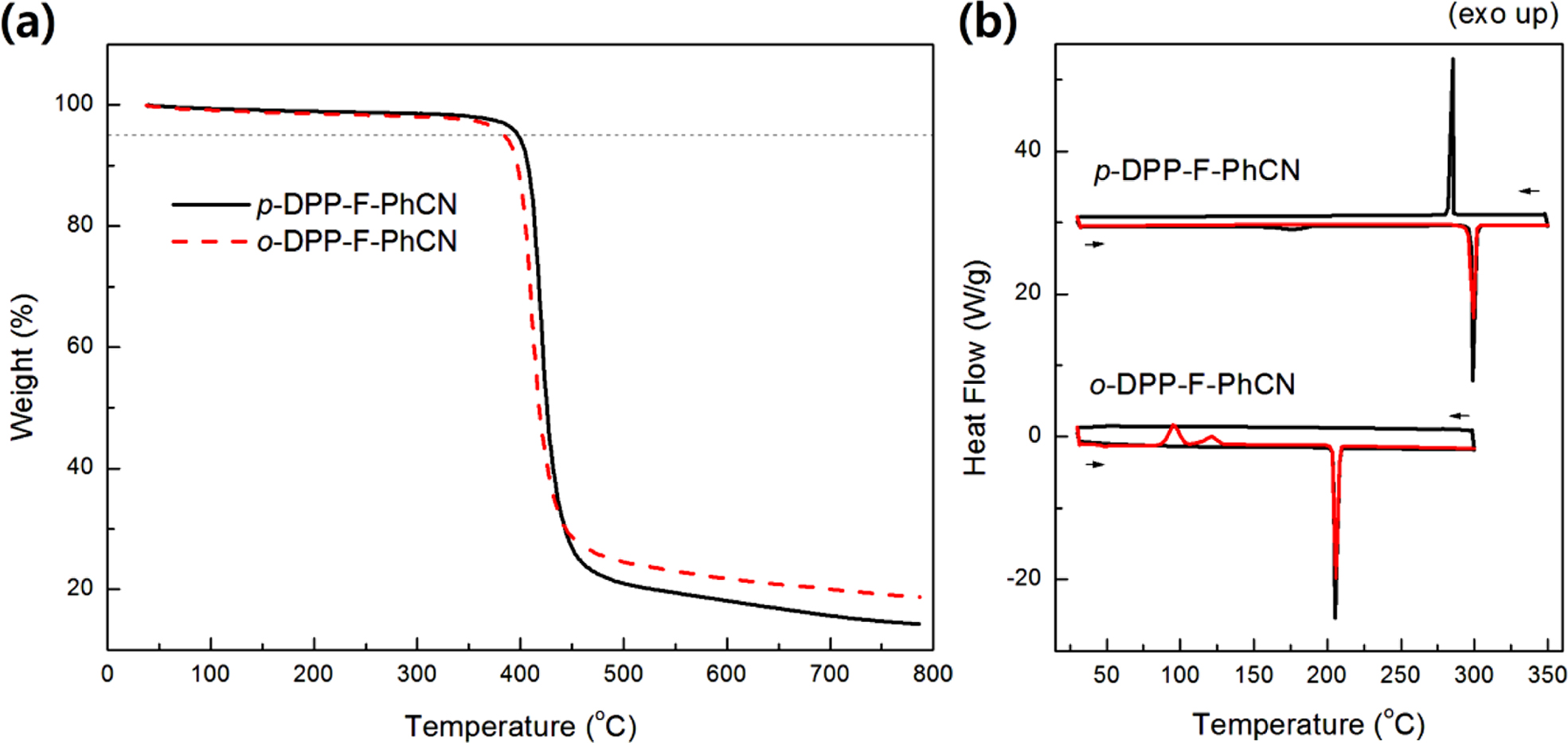
The thermal characteristics of the small molecules are summarized in
Table 1. The two compounds were thermally stable, showing 5% weight losses upon
being heated to 380 °C in TGA analysis (Figure 1(a)). The phase-transition
temperatures were measured by DSC (Figure 1(b)). The melting temperatures (Tm)
of p- and o-DPP-F-PhCN were 300 and 206 oC,
respectively. We likewise observed a higher Tm for the
para-substituted analogue in our previous work with DPP-PhCNs,12
where p-DPP-PhCN exhibited a higher Tm (251 °C)
than o-DPP-PhCN (Tm = 187 °C).
In addition, the introduction of furan resulted in higher Tm
values; for example, the Tm of p-DPP-F-PhCN
(300 °C) was higher than that of the thiophene analogue p-DPP-PhCN
(Tm = 251 °C) by approximately 50 °C.
Notably, o-DPP-F-PhCN exhibited an enthalpy change (ΔHm
= −100 J/g) more negative than that of p-DPP-F-PhCN (−85 J/g),
reflecting the relatively better crystallinity of o-DPP-F-PhCN. By
contrast, in our previous work on DPP-PhCNs, p-DPP-PhCN exhibited a ΔHm
(−111 J/g) more negative than that of o-DPP-PhCN (−86 J/g).12
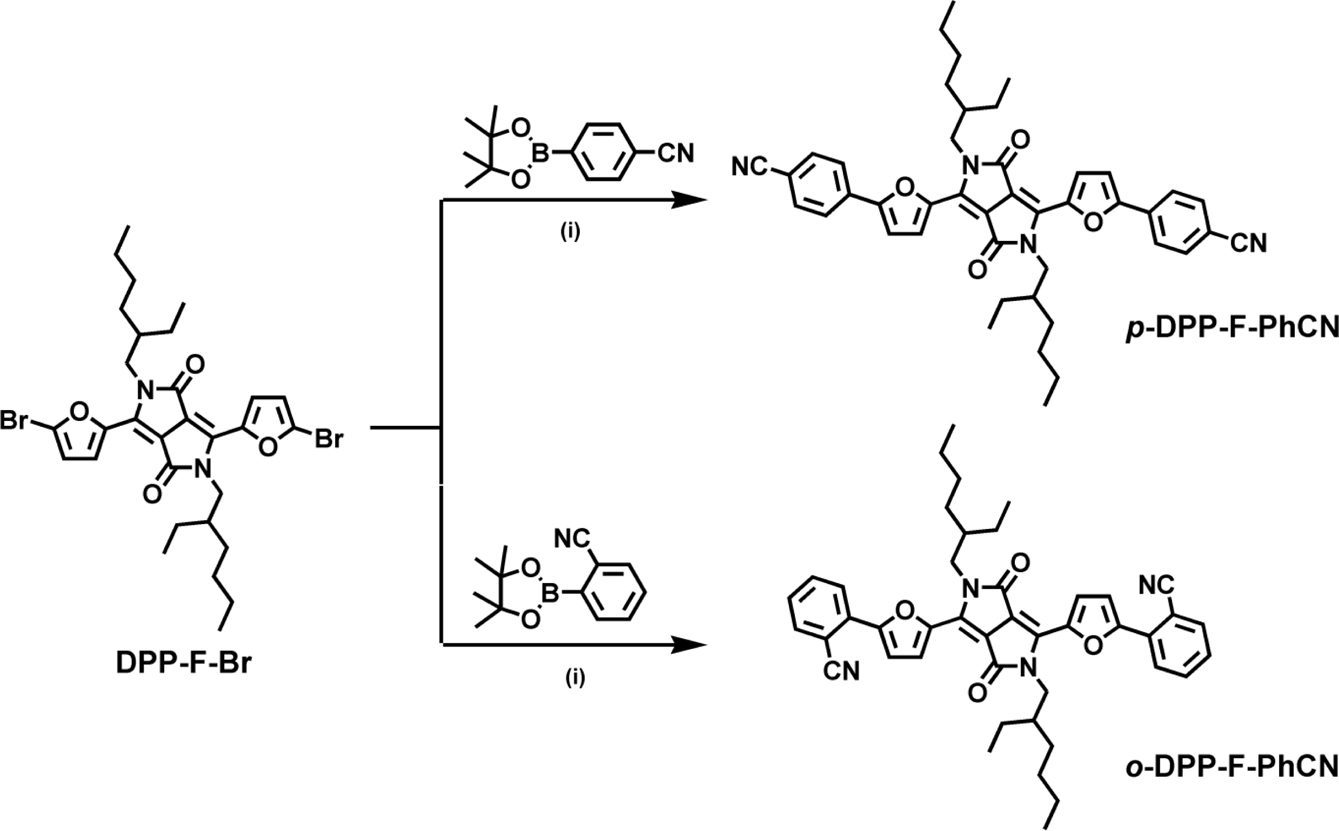
Scheme 1. Synthetic procedure for DPP-F-PhCNs. (i) Pd2(dba)3, P(t-Bu)3 × HBF4, K3PO4, THF, H2O, 80 °C, 12 h.
Optical
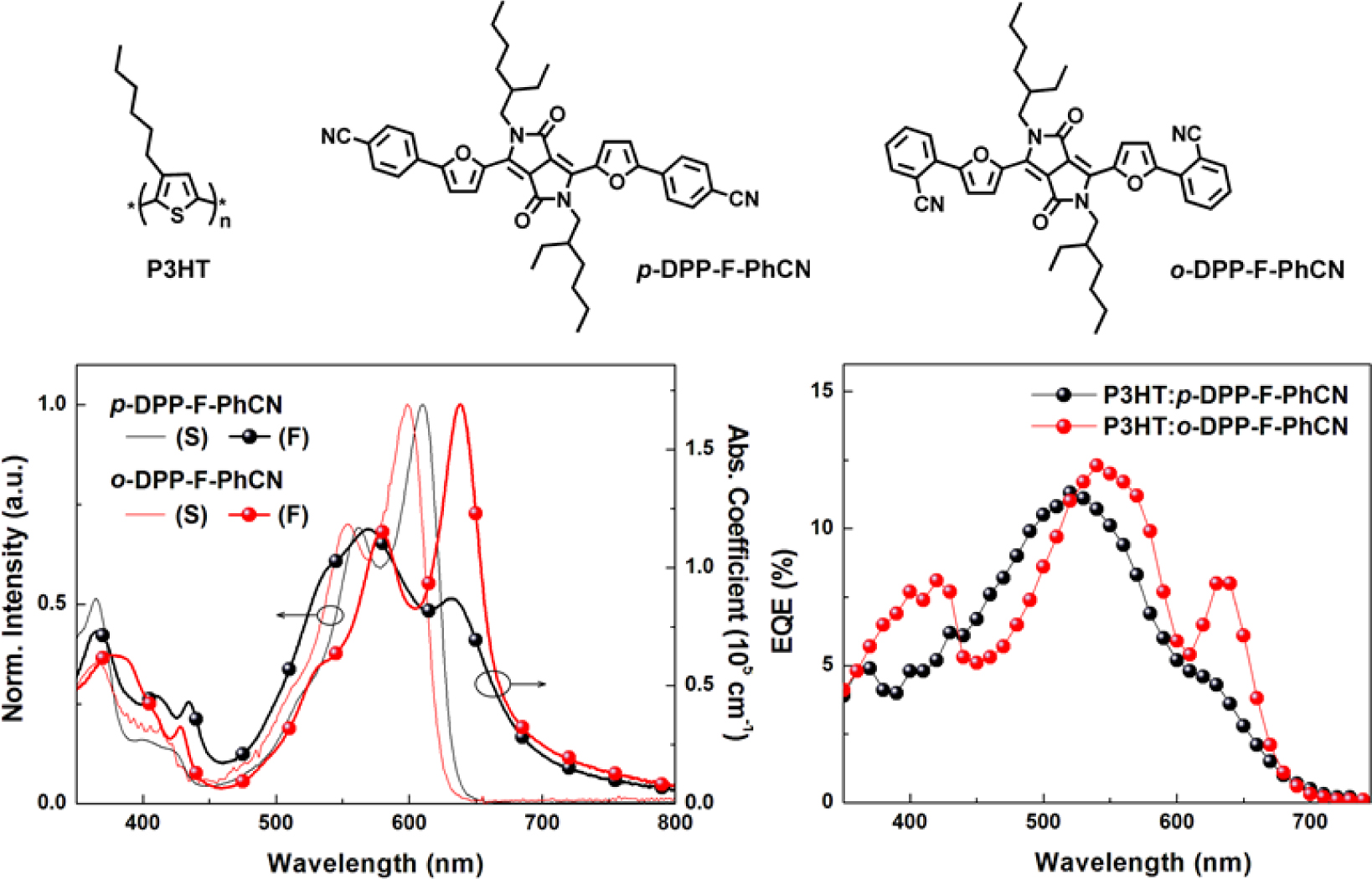
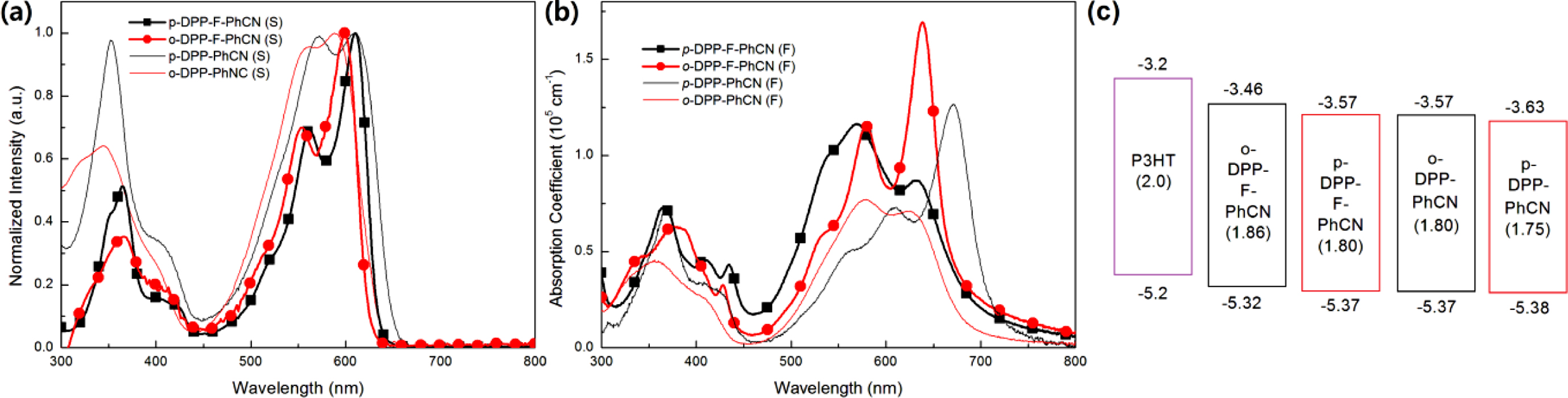
and Electrochemical Properties. The UV–vis absorption spectra
of p- and o-DPP-F-PhCN in the solution and film states are shown
in Figure 2. In the solution state, p- and o-DPP-F-PhCN exhibited
similar UV–vis absorption profiles; however, the absorption maximum of p-DPP-F-PhCN
(lmax = 601 nm) was slightly red-shifted compared with
that of o-DPP-F-PhCN (lmax = 599 nm), in
agreement with the trend observed for DPP-PhCNs. That is, the conjugation in
the para-substituted analogue might be slightly longer than that in the ortho-substituted
analogue. In the spectra of p- and o-DPP-F-PhCN films, the
absorption peaks were broader than those in the spectra of the solution state
samples. In particular, as shown in Figure S3, the absorption maximum of o-DPP-F-PhCN
was dramatically red-shifted by approximately 40 nm from its position in the
solution-state spectrum (lmax = 599 nm) to that
in the film-state spectrum (lmax = 638 nm). We
observed a similar phenomenon in our previous experiments with p-DPP-PhCN
(but not o-DPP-PhCN),12 where the greater red-shift in the
absorption of p-DPP-PhCN (lmax = 670 nm) was
explained by its high degree of molecular aggregation. Interestingly, the film
aggregation behavior and absorption profiles of the small molecules used in the
present study were controlled by the nature of the p-bridge (i.e.,
thiophene vs. furan) as well as by the position of the substituents (i.e.,
para vs. ortho). In addition, the optical bandgaps (Eg,opt)
of the small molecules were calculated to be 1.80 and 1.86 eV for p- and
o-DPP-F-PhCN, respectively, using the following equation: Eg,opt = 1240/lonset (eV), where lonset is the wavelength
of the absorption onset of the small molecule films.
The highest occupied molecular orbital (HOMOelec) and the
lowest unoccupied molecular orbital (LUMOelec) energy levels were
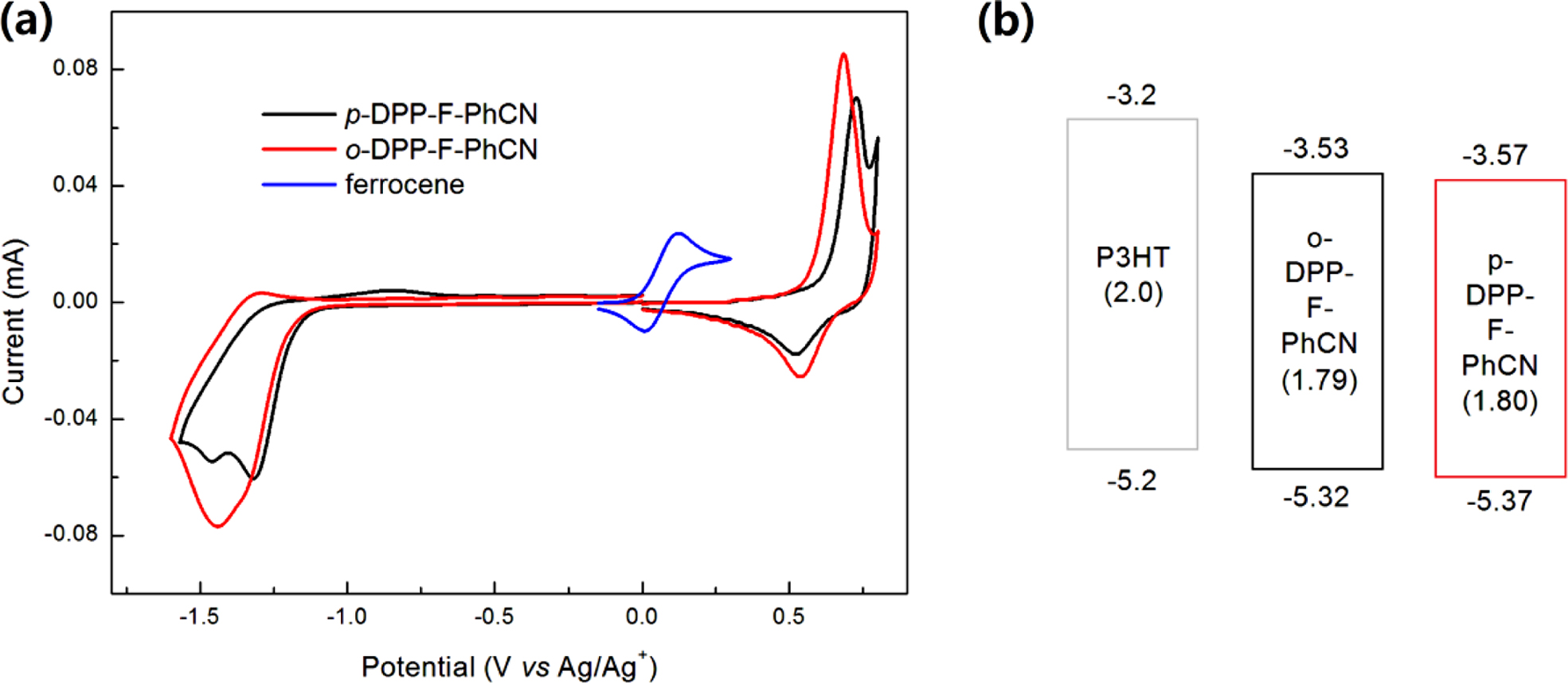
estimated from the CV, as shown in Figure 3, using the following equations:
HOMOelec = (Eonset,ox − E1/2,ferrocene + 4.8)
eV and LUMOelec = (Eonset,red − E1/2,ferrocene + 4.8)
eV, where Eonset,ox and Eonset,red are the onset
potentials of oxidation and reduction, respectively, assuming that the energy
level of ferrocene (Fc) is 4.8 eV below the vacuum level.6,16-18 The
position of the substituent (CN) affects the energy levels of the small
molecules: para-substitution resulted in relatively low-lying HOMOelec/LUMOelec
energy levels for p-DPP-F-PhCN (−5.37/−3.57 eV) relative to those of o-DPP-F-PhCN
(−5.32/−3.53 eV). Both small molecules have sufficient HOMO and LUMO offsets
with respect to the P3HT polymer used as a donor in PSC fabrication, enabling
not only electron transfer from the P3HT to the small molecules but also hole
transfer from the small molecules to the P3HT (i.e., Channel II charge
generation).12
Figure 2(c) compares the energy levels of DPP-F-PhCNs with those of the
DPP-PhCN thiophene analogues to illustrate how the incorporation of the furan
moiety affects the electrochemical properties of the small molecules; the
optical LUMO energy levels (LUMOopt) were estimated from the HOMOelec
energy levels and the optical Eg,opt (LUMOopt =
HOMOelec + Eg,opt). The furan
substitution elevated both the HOMO and LUMO energy levels of the small
molecules, where it exerted a slightly stronger effect on the LUMO levels than
on the HOMO levels.
Photovoltaic
Performances. PSCs were fabricated with the configuration ITO/PEDOT:PSS/active
layer/LiF/Al. Both small molecules could act as acceptors when combined with
regioregular P3HT as a polymer donor because of the sufficient HOMO and LUMO
offsets between the donor and acceptors, as previously discussed (Figure 3). Moderate
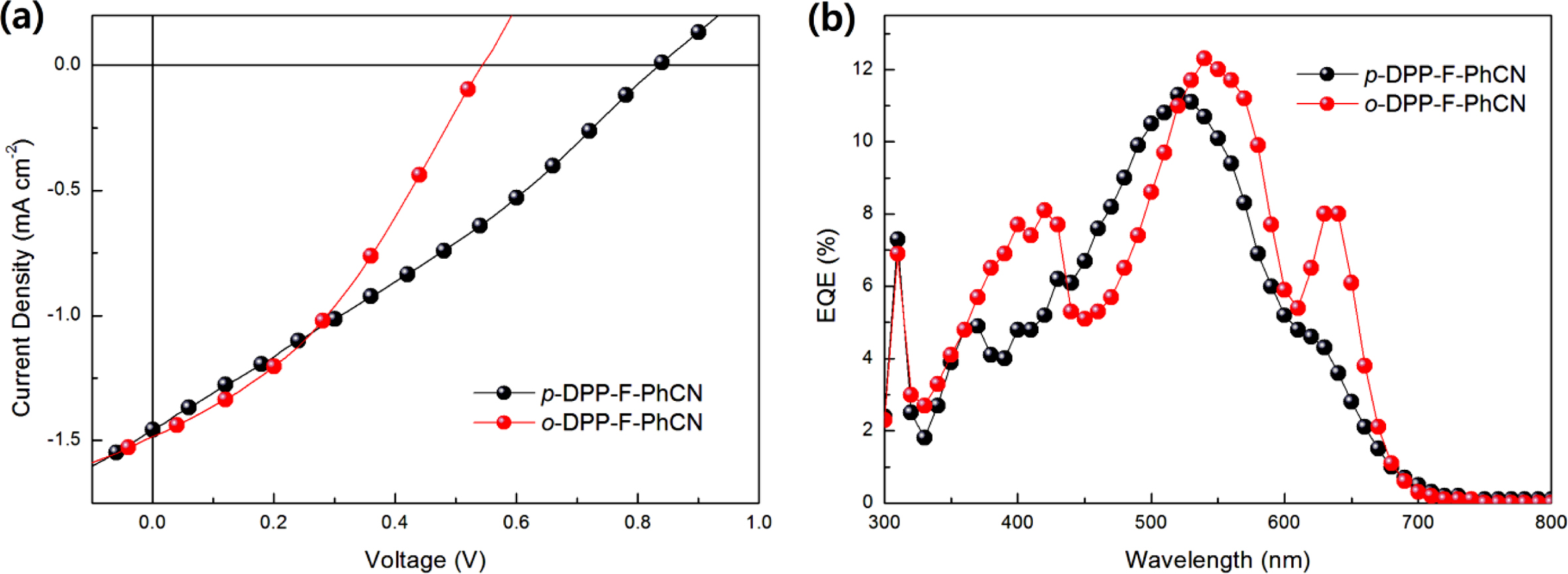
PCEs of ~0.3% were obtained with the P3HT:DPP-F-PhCN devices. The J–V
and EQE curves were recorded in air under white-light AM 1.5G illumination
(100 mW cm−2) and are shown in Figure 4. The active area (i.e.,
the aperture area in a mask) was defined as 9 mm2. The optimized
photovoltaic properties are summarized in Table 2.
The short-circuit current density (JSC) values were
found to be similar between the devices with the two DPP-F-PhCNs as well as
between the devices with the two DPP-PhCNs. The maximum EQE intensities of the
DPP-F-PhCN-based devices were also similar; however, we note that the EQE
profile of the o-DPP-F-PhCN-based device was red-shifted relative to
that of the p-DPP-F-PhCN-based device, consistent with the absorption
profiles of the two DPP-F-PhCN films. In particular, the additional sharp EQE
response at 635 nm coincides with the absorption maximum of the o-DPP-F-PhCN
film (λmax = 638 nm). Interestingly, the EQE and
absorption profiles of o-DPP-F-PhCN are similar to those of p-DPP-PhCN,
not those of o-DPP-PhCN. Among the thiophene analogues of DPP-PhCNs, p-DPP-PhCN
exhibited the highest degree of molecular aggregation, whereas the introduction
of a furan moiety altered the aggregation behavior of the molecules and the ortho-substitution
resulted in greater molecular aggregation and a red-shift of the absorption
maximum, leading to the additional EQE response at long wavelengths.
The slightly higher PCE of p-DPP-F-PhCN originated from its higher
open-circuit voltage (VOC) of 0.83 V compared with that of o-DPP-F-PhCN
(VOC = 0.54 V). This result is inconsistent
with the general understanding that the VOC values of the
devices generally depend on the energy difference between the HOMO level of the
donor and the LUMO level of the acceptor.19 For a given HOMO level
of the donor, o-DPP-F-PhCN, which has a higher LUMO level than p-DPP-F-PhCN,
is expected to result in a device with a higher VOC; however,
the VOC of the o-DPP-F-PhCN-based device (VOC = 0.54
V) was lower than expected. This result is attributable to the strong
aggregation of o-DPP-F-PhCN, as confirmed by its absorption spectrum
(Figure 2) and high DHm, where the
enhanced orbital interactions could destabilize/stabilize the HOMO/LUMO levels
of the aggregates.12 Further improvements of photovoltaic properties
could be achieved by optimizing device fabrication conditions and introducing a low
bandgap polymer donor or a third component for ternary cells.)
|
Figure 1 (a) TGA; (b) DSC curves of DPP-F-PhCNs. |
|
Figure 2 UV absorption spectra in (a) solution; (b) films; (c) energy diagram of DPP-F-PhCNs and DPP-PhCNs. The values in parentheses are Eg,opt, which were calculated from the absorption onset of the small molecule films (Eg = 1240/λonset (eV)); the LUMOopt levels were calculated from the HOMOelec and Eg,opt values. |
|
Figure 3 (a) CV; (b) energy diagrams of DPP-F-PhCNs. The values in parentheses are the electrochemical energy bandgaps (Eg,elec) calculated from the HOMOelec and LUMOelec energy levels in CV measurements. |
|
Figure 4 (a) J-V curves; (b) EQE responses of P3HT:acceptor devices. |
|
Table 1 Physical Properties of DPP-F-PhCNs |

aT5d are the decomposition temperatures showing 5% of the weight losses. bEg,opt were calculated from the onset of absorption spectra in film (Eg,opt=1240/λonset (eV)). cLUMOopt were estimated using HOMOelec levels and Eg,opt (LUMOopt= HOMOelec+ Eg,opt). |
|
Table 2 Optimized Photovoltaic Performances of P3HT: Acceptor Devicesa |
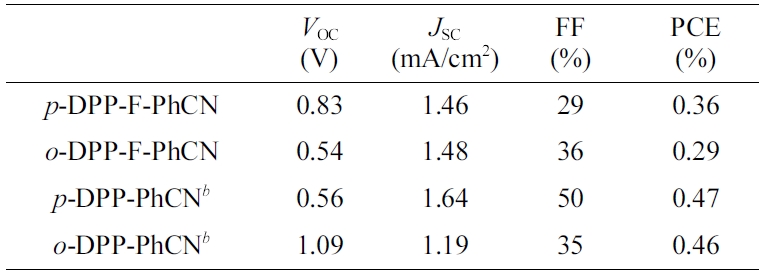
aThe device architecture is ITO/PEDOT:PSS/P3HT:acceptor (1:1, w/w)/LiF/Al. The active layers were prepared by spin-coating of a chloroform solution of donor and acceptor, and the devices were annealed for 10 min. bData taken from ref [12]. |
The p- and o-DPP-F-PhCN were synthesized for use as NFAs in
PSCs. The optical and electrochemical properties of the small molecules varied
according to both the substituent position and the furan effect. Among the
DPP-F-PhCNs and DPP-PhCNs, the highest-lying HOMO and LUMO levels were observed
in o-DPP-F-PhCN because of both the furan introduction and the
ortho-substitution. The relatively red-shifted and enhanced λmax
in the UV–vis absorption spectra and greater enthalpy change of o-DPP-F-PhCN
relative to that of p-DPP-F-PhCN indicated stronger molecular
aggregation behavior of o-DPP-F-PhCN, leading to the observed EQE
response at long wavelengths. Such strong aggregation of o-DPP-F-PhCN
resulted in a relatively low VOC value in the device despite
its highest-lying LUMO level, which can be explained by the enhanced orbital
interactions and thus stabilized LUMO level of o-DPP-F-PhCN.
- 1. R. D. Maduwu, H. C. Jin, and J. H. Kim, Macromol. Res., 27, 1261 (2019).
-
- 2. M. H. Hoang, G. E. Park, D. L. Phan, T. T. Ngo, T. V. Nguyen, C. G. Park, M. J. Cho, and D. H. Choi, Macromol. Res., 26, 844 (2018).
-
- 3. L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao, and Y. Chen, Science, 361, 1094 (2018).
-
- 4. Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Tang, B. Lin, K. Xian, B. Gao, C. An, P. Bi, W. Ma, and J. Hou, Nat. Sci. Rev., nwz200 (2019).
-
- 5. S. Pang, X. Zhou, S. Zhang, H. Tang, S. Dhakal, X. Gu, C. Duan, F. Huang, and Y. Cao, ACS Appl. Mater. Interf., 12, 16531 (2020).
-
- 6. T. Lee, Y. Eom, C. E. Song, I. H. Jung, D. Kim, S. K. Lee, W. S. Shin, and E. Lim, Adv. Energy Mater., 9, 1804021 (2019).
-
- 7. X. Li, Y. Wang, Q. Zhu, X. Guo, W. Ma, X. Ou, M. Zhang, and Y. Li, J. Mater. Chem. A, 7, 3682 (2019).
-
- 8. Z.-P. Yu, Z.-X. Liu, F.-X. Chen, R. Qin, T.-K. Lau, J.-L. Yin, X. Kong, X. Lu, M. Shi, C.-Z. Li, and H. Chen, Nat. Commun., 10, 2152 (2019).
-
- 9. T. Liu, X. Pan, X. Meng, Y. Liu, D. Wei, W. Ma, L. Huo, X. Sun, T. H. Lee, M. Huang, H. Choi, J. Y. Kim, W. C. H. Choy, and Y. Sun, Adv. Mater., 29, 1604251 (2017).
-
- 10. V. S. Gevaerts, E. M. Herzig, M. Kirkus, K. H. Hendriks, M. M. Wienk, J. Perlich, P. Müller-Buschbaum, and R. A. J. Janssen, Chem. Mater., 26, 916 (2013).
-
- 11. E. Lim, S. Lee, K. K. Lee, I.-N. Kang, S.-J. Moon, H.-Y. Kong, and H. E.Katz, Sol. Energ. Mat. Sol. C, 107, 165 (2012).
-
- 12. Y. Kim, C. E. Song, E. J. Ko, D. Kim, S. J. Moon, and E. Lim, RSC Adv., 5, 4811 (2015).
-
- 13. C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee, and J. M. J. Fréchet, J. Am. Chem. Soc., 132, 15547 (2010).
-
- 14. P. Sonar, S. P. Singh, E. L. Williams, Y. Li, M. S. Soh, and A. Dodabalapur, J. Mater. Chem., 22, 4425 (2012).
-
- 15. H. Bürckstümmer, A. Weissenstein, D. Bialas, and F. Würthner, J. Org. Chem., 76, 2426 (2011).
-
- 16. Y. Eom and E. Lim, Polym. Korea, 39, 986 (2015).
-
- 17. D. Liu, B. Kan, X. Ke, N. Zheng, Z. Xie, D. Lu, and Y. Liu, Adv. Energy Mater., 8, 1801618 (2018).
-
- 18. M. Zhang, Z. Xiao, W. Gao, Q. Liu, K. Jin, W. Wang, Y. Mi, Q. An, X. Ma, X. Liu, C. Yang, L. Ding, and F. Zhang, Adv. Energy Mater., 8, 1801968 (2018)
-
- 19. M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger, and C. J. Brabec, Adv. Mater., 18, 789 (2006).
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2020; 44(3): 408-414
Published online May 25, 2020
- 10.7317/pk.2020.44.3.408
- Received on Mar 25, 2020
- Revised on Apr 13, 2020
- Accepted on Apr 14, 2020
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
- Supporting Information
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Eunhee Lim
-
Department of Chemistry, Kyonggi University, 154-42 Gwanggyosan-ro, Yeongtong-gu, Suwon 16227, Korea
- E-mail: ehlim@kyonggi.ac.kr
- ORCID:
0000-0002-2321-7072
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr