






- Thiol-Michael Coupling and ROMP: Comparison of the Effects of Steric Hindrance with Functional Dendronized Polymer
Tong Wu†
 , Maojie Xuan, Xingyou Wang, and Meina Liu†
, Maojie Xuan, Xingyou Wang, and Meina Liu† School of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai, 100 Haiquan Road, China
- 메르캅탄(Mercaptan)-마이클 결합(Michael Coupling)과 ROMP: 기능성 나뭇가지형 폴리머의 공간 방해 효과에 대해 비교
Dendronized
exo-7-oxanorbornene monomers were synthesized and polymerized via thiol–Michael
coupling and ring-opening metathesis polymerization. The polymerization rate was highly dependent on the steric hindrance of the terminal group. The space linker between the polymerizable group and
dendron was a crucial factor. Ru-based kinetics was investigated using nuclear magnetic resonance (NMR), which showed that the monomer was completely converted to a
generally narrow polydispersity material. The second generation of functional
macromonomer was grafted from the first with a different linker length and consumed completely at 50 oC in
toluene. Considering the properties of the space linker, we found that a short space linker of dendronized exo-7-oxanorbornene macromonomer leads to narrow molecular weight distributions of obtained dendronized
polymers. This work provides a further understanding of the effect of steric hindrance
on dendronized polymers, thereby allowing the development of functional
materials.
Comparing the effects of steric hindrance by making
well-defined dendronized polymer with different space linker and different
functional groups when incorporating bifunctional group to the polymer. This is
positive for the acquisition of functional dendronized polymer.

Keywords: thiol–Michael, ring-opening metathesis polymerization, functional polymer, steric hindrance
This work was supported by the National Natural
Science Foundation of China (No. 21604056), Natural Science Foundation of
Shanghai (No. 16ZR1435600), State Key Laboratory of Molecular Engineering of
Polymers (No. K2017-16), Key Laboratory of Synthetic, and Self-Assembly Chemistry
for Organic Functional Molecules (No. K2017-7).
Dendronized polymers, as important topological structures of branched
polymer, have exhibited impressive advantages in terms of biology, medicine,
catalysis, nanomaterials, and photoelectric materials in recent years.1
Moreover, the highly branched and regular structures of dendronized polymers
have unique physical and chemical properties, such as high rheological properties,
good solubility and several modifiable terminal functional groups. These polymers are generally obtained from dendronized motifs through
convergent and divergent methods.2,3 However, the
synthesis process is immensely tedious that the deprotection
and activation steps between each generation of dendronized polymers are
required.4,5 Recently, considerable effort has been
devoted to macromonomer route research. Considering the
facile and streamlined approach, the dendronized monomer with polymerizable groups is synthesized and polymerized
via living polymerization.
Living polymerization is an efficient measure for realizing molecular
design, and synthesizing a series of polymer materials with different
structures and properties. Effective strategies such as
atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer polymerization (RAFT), and ring-opening metathesis polymerization (ROMP), have been developed to design
synthetic dendronized polymers. During ATRP, the removal process of transition
metal complexes in polymers was cumbersome.6,7
Simultaneously, disulfide derivatives in RAFT may increase the toxicity of
polymers.8 However, the initiation/propagation
rate (ki/kp) ratio of ROMP increases with the catalyst development.9,10 Importantly, the degree of polymerization (DP) has a completely linear relationship with the consumed monomer.11 Furthermore, the use of various
thiol-based chemistries as tools for complex molecule synthesis,
polymerization, and post-polymerization modification has received
significant interest.12-14 For example,
exclusively low levels of phosphine have been used as initiating species to catalyze the formation of the thiol–Michael adduct rapidly. On one hand, this approach can synthesize dendronized polymers
with appropriate molecular weight distribution and high algebraic distribution;
on the other hand, it can combine with “click” chemistry to produce multiple
functional dendronized polymers.15-18
Recently, dendronized polymers have attracted significant attention due to their unique properties,
specifically with the development of highly
efficient active ROMP catalysts (Grubbs catalysts) and the diversification of
polymerized monomers (cyclic olefins extended to cyclic monomers containing
heteroatoms and polar functional groups). ROMP technology can prepare a dendronized polymer with controllable molecular weight via a
macromolecular route.19 Schlüter et al.
first used ROMP technology to prepare a dendronized polymer with a molecular
weight of 216000 with RuCl3 as catalyst.20 With the limitation of RuCl3, the polymerization reaction did
not exhibit the characteristic of living polymerization. Then, the same group reported the first case of active ROMP reaction. With
RuCl2(=CHPh)(PCy3)2 as catalyst, they obtained
a series of dendronized polymers with a molecular weight of 86900 by adjusting
the ratio of the initial monomer and catalyst.21
Fréchet
et al. successfully prepared high molecular weight dendronized polymers with biphenyl as the linking group containing
second- and third-generation dendronized polymers with ruthenium-based catalyst, they confirmed dendronization through atomic force
microscopy imaging.22 Weck and coworker found that an effective linker between the polymerizable group and the dendron can
remarkably increase the polymerization rate.23 Thus, as the
degree increases, the monomer molecular chain elongates, and the terminal
structure enlarges. These two factors have a potential effect during polymerization and an indispensable effect on polymer materials.
Herein, difunctional and tetrafunctional branched dendronized
polymers were prepared via thiol–Michael coupling and ROMP. Among them,
second-generation dendronized aromatic monomers were completely consumed in
toluene at an elevated temperature and a topological material was
obtained (Mn = 23003, PDI = 1.31). The
steric hindrance effect observed by NMR is evident during
polymerization. As the end structure enlarges, the polymerization rate slows
down, and further severe conditions are required to convert it
fully. The space linker effect is also reflected. When the molecular chain
shortened, we obtained dendronized polymers with a narrow PDI. The selected structures were used to produce functional materials for
determining their polymerization performance.
Materials. Anhydrous
methanol, dichloromethane and tetrahydrofuran (THF) were purchased
from Macklin Inc. Anhydrous toluene and triethylamine were used after
redistillation. Other reagents were purchased from the Tansoole Co., Ltd (Shanghai, China) at the highest purity
and used as received, unless noted otherwise.
Instrumentation. NMR spectra were
recorded on a Bruker AVANCE III spectrometer at 500 MHz for hydrogen nuclei and
125 MHz for carbon nuclei in an appropriate deuterated solvent. Chemical shifts
(δ) for 1H NMR spectra were reported in parts per million (ppm).
Data were presented as follows: chemical shift (multiplicity: s, singlet; d,
doublet; dd, doublet of doublets; t, triplet; m, multiplet), coupling constants in Hz, and integration. THF
size-exclusion chromatography (SEC) analyses were performed on a Shimadzu
modular system comprising an autoinjector and a 5.0 mm bead-size guard column (50 × 7.5 mm2) from Polymer Laboratories, followed by three linear PL columns and a
differential refractive index detector using THF as the eluent at 40 oC
with a flow rate of 1 mL min−1. The system was calibrated with
abovementioned linear polystyrene standards with narrow molecular weight
distribution. High-resolution mass spectra (HRMS) were acquired on a Bruker
Solaril X70 spectrometer with an Anakytica source.
Synthesis
of 2-ene and 4-ene. Seven compounds
were synthesized in good yields and characterized by NMR and HRMS (Scheme 1).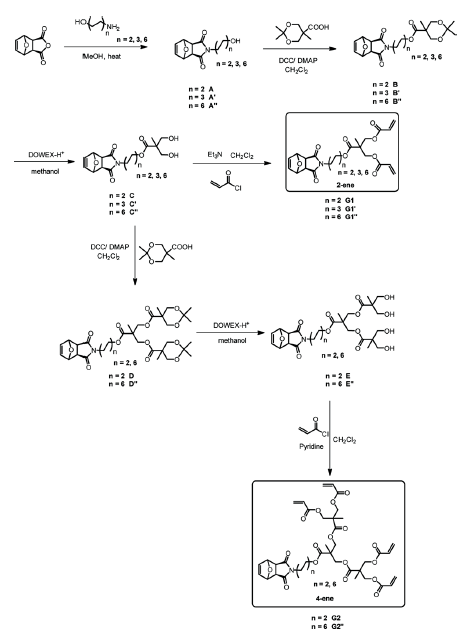
Scheme 1. Outline for the preparation of target 2- and 4-functionalacrylic exo-7-oxanorbornene dendron monomers.
Synthesis
of thiol–Michael Adducts. Table 1 shows a
typical procedure for the thiol–Michael addition of a thiol, namely, R-SH, to
2-ene. A scintillation vial (20 mL capacity) was added with 2-ene (1.0 mmol, 1.0 equiv.) and the desired thiol (2.1 equiv.) dissolved
in CH2Cl2 (2.0 mL). Dimethylphenylphosphine (Me2PPh;
5 mol%) was then added to the solution and the reaction was allowed to
proceed at room temperature. The reaction was monitored by TLC and halted when
the reagents were completely consumed. Solvent was removed under reduced
pressure to provide the crude Michael adducts. Pure products were obtained
after purification through flash chromatography. Reported yields were an
average of at least two runs.
Table 2 presents a typical procedure for the
thiol–Michael addition of a thiol, namely, R-SH, to 4-ene. A scintillation vial (20 mL) was added with 4-ene (1.0 mmol,
1.0 equiv.) and the desired thiol (5.0 equiv.) dissolved in CH2Cl2
(2.0 mL). Me2PPh (8 mol%) was then added to the solution, and the reaction was allowed to proceed at room temperature. The
reaction was monitored by TLC and halted when the reagents were completely
consumed. Solvent was removed under reduced pressure to provide the crude
Michael adducts. Pure products were obtained after purification through flash
chromatography. Reported yields were an average of at least two
runs.
2-(((6-((3aR,7aS)-1,3-dioxo-1,3,3a,4,7,7a-hexahy-dro-2H-4,7-epoxyisoindol-2-yl)hexyl)oxy)carb-nyl)-2-methylpropane-1,3-diylbis(3-((3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)thio) propanoate) (M3)
G1’’ (0.122 g, 0.25 mmol) and 1H,1H,2H,2H-perfluorodecanethiol
(121 μL, 0.525 mmol) were reacted for 3 h. The Michael
adduct was isolated as a white solid in an 83% yield (0.26
g). 1H NMR (500 MHz, CDCl3, ppm) δ = 6.52 (s, 2H), 5.27
(s, 2H), 4.28 (dd, J = 25.5, 11.1 Hz, 4H), 4.12 (t, J = 6.5 Hz, 2H), 3.48 (t, J
= 7.2 Hz, 2H), 2.86–2.73 (m, 10H), 2.64 (t, J = 7.1 Hz, 4H), 2.41 (dt, J =
26.5, 9.0 Hz, 4H), 1.66–1.55 (m, 4H), 1.41–1.31 (m, 4H), 1.27 (s, 3H). 13C
NMR (125 MHz, CDCl3, ppm) δ = 176.22, 172.49, 170.96, 136.43, 122.79–106.15 (m,
CF2, CF3), 80.86, 65.41, 65.06, 47.31, 46.16, 38.56, 34.26,
31.98, 31.82, 31.64, 29.59, 28.20, 27.28, 26.89, 25.99, 25.19, 22.64, 17.77. 19F
NMR (470 MHz, CDCl3, ppm) δ = −80.80 (m, 6F), −114.34 (m, 4F), −121.40
to −124.25 (m, 12F), −126.16 (m, 4F). HRMS: calculated for C41H41F26NO9S2
[M+H+] 1249.8555, found 1249.8557.
(((2-(((6-((3aR,7aS)-1,3-dioxo-1,3,3a,4,7,7a-hexahydro-2H-4,7-epoxyisoindol-2-yl)hexyl)oxy)
carbonyl)-2-methylpropane-1,3-diyl)bis(oxy))bis (carbonyl))bis(2-methylpropane-2,1,3-triyl)
tetrakis(3-(benzylthio)prop-anoate)
(M23)
G2’’ (0.913 g, 1.1 mmol) and benzyl mercaptan (646 μL, 5.5 mmol) were
reacted for 6 h. The adduct was isolated as a colorless syrup in
a 60% yield (0.825 g). 1H NMR (500 MHz, CDCl3, ppm) δ =
7.28 (d, J = 31.0 Hz, 20H), 6.51 (s, 4H), 5.26 (s, 4H), 4.26 (d, J = 3.2 Hz,
26H), 4.10–4.08 (m, 5H), 3.72 (d, J = 1.4 Hz, 18H), 3.47 (s, 5H), 2.83–2.80 (m,
14H), 2.65 (d, J = 5.1 Hz, 8H), 1.62–1.56 (m, 10H), 1.24 (s, 11H). 13C
NMR (125 MHz, CDCl3, ppm) δ = 176.29, 172.09, 171.25, 169.70,
138.01, 136.54, 128.85, 128.56, 127.11, 80.92, 65.70, 65.46, 65.17, 47.39, 46.59,
46.36, 38.88, 38.69, 37.34, 36.24, 34.23, 28.32, 27.36, 26.12, 25.32, 17.84.
HRMS: calculated for C69H83NO17S4 [M+H+]
1326.6531, found 1326.6536.
Typical
Procedure for the Online Monitoring of ROMP of the Thiol–michael Adducts via 1H NMR Spectroscopy. In 20 mL glass
bottles, monomers and catalysts (Grubbs, the ratio being
dictated by the targeted molecular weight) were dissolved in
solvent and then stirred under nitrogen protection. The polymerization inhibitor (ethyl vinyl ether) was
added to
the solution and stirred for 30 min. The conversion was monitored
by comparing the ratio of the integrals of the polymeric to monomeric ene peaks
versus time with NMR.


Thiol–Michael Coupling. We selected
poly(exo-7-oxanorbornene) as the polymerizable backbone and a polyester-based
structure. Exo-7-oxanorbornene was linked to the backbone with various lengths
of carbon chains. Thiol was grafted onto the end of the branched
macromolecule via Michael addition of thiol olefins to obtain functional
monomers. This work aimed to compare the effect of steric hindrance with
functional groups. We initially targeted the difunctional and
tetrafunctional derivatives of diacrylate and tetraacrylate functional dendron
species that would serve as common substrates in ROMP.
As outlined in Scheme 1, 2-ene and 4-ene were prepared via multistep
procedures. They were synthesized in a four-step process involving the initial
preparation of 2,2,5-trimethyl-1,3-dioxane-5-carboxylic acid that was
subsequently reacted with alcohol A’’ in a carbodiimide coupling to
provide the protected diol B’’. Treatment of B’’ with Dowex-H+ generated the corresponding free diol C’’. C’’
serves as a common precursor to G1’’ and G2’’ because it can be
acylated with acryloyl chloride, to produce G1’’ or reacted with
2,2,5-trimethyl-1,3-dioxane-5-carboxylic acid in another carbodiimide-mediated
esterification to provide protected tetraol D’’, which generates
free tetraol E’’ after treatment with Dowex-H+.
Esterification of tetraol (E’’) with acryloyl chloride, mediated by
pyridine, provides target tetraacrylate G2’’.24 Pyridine has excellent performance in esterifying multiple
sites. In all instances, the yields of various
synthetic steps were good to excellent, and all products were
characterized using standard methods. As an example, Figure 1 shows the 1H NMR spectrum, recorded in CDCl3, of
diacrylate dendron monomers G1’’ and G2’’ with peak
assignments confirming their structure.
2-ene and 4-ene converted numerous thioether adducts via Me2PPh-mediated
thiol–Michael coupling reactions (Tables 1 and 2,
respectively).25 Seven mercapto
compounds were 1H, 1H,2H,2H-perfluorodecanethiol, the siloxy
species 3-mercaptopropyltriethoxysilane, benzyl mercaptan, methyl 3-mercaptopropionate, mercaptopropyl-isobutyl-POSS, 3-mercapto-1-propanol, and acetyl-thiol-glucose. In all
instances, the target adducts were isolated in good to
excellent yields, and their structures were confirmed using a combination of NMR spectroscopy and HRMS. In addition, the
tetrafunctional–aromatic adduct was prepared from the reaction
of 4-ene with benzyl mercaptan. The tetrafunctional thioether adduct was
isolated in a 60% yield. However, other thiols have a Michael
addition reaction with 4-ene, leading to the low isolated yield. As a
representative example, Figure 2 shows the 1H NMR
spectrum for silicone functional dendron species M12. Several points are
worth highlighting. Importantly, the alkane
resonance associated with methyl was clearly visible at δ = 1.21 ppm
(labelled h). A simple ratio of these signals
with a or b (the vinylic Hs associated with the oxanorbornene and the
corresponding allylic Hs at the bridgehead O) confirmed the structure. In
addition, we observed two sets of peaks at δ = 2.74 and 2.54 ppm, which were
closely related to the bridging of the bridged sulfur bonds between the
siloxane functional groups and olefins. All peaks integrated in a ratio
expected for the target structure.
ROMP. With a small library of
thioether functional 2-ene derivatives available, we evaluated the capability
to homopolymerize substrates with Grubbs’ [Ru]-based ROMP initiators. In initial screenings, we intentionally
targeted low molecular weight homopolymers (target Mn = 10 kDa)
to determine the capability of the highly functional and sterically bulky
monomers to undergo polymerization. Figure 3 shows the representative 1H
NMR and kinetic conversion data for the homopolymerization (target Mn = 10 kDa)
of the dendron monomers derived from difunctional derivative.
The reaction in Figure 3(A) shows the homopolymerization of the aromatic
dendron monomer M8. This example indicates that the double bond of the 7-oxanorbornene was broken to form a chain structure under the Ru-based catalysis. Figure
3(B) presents a series of selected 1H NMR spectra, each of which was
directly recorded in the spectrometer for the homopolymerization of the aromatic
dendron monomer. 1H NMR spectroscopy is a convenient
technique that can be applied following monomer consumption in ROMP. This fact stems from the distinct chemical shift of the vinyl resonance
associated with the monomer, compared with the cis/trans vinylic bonds
in the resulting polymer.26,27 We can clearly
observe the monomer vinyl resonance above δ = 6.5 ppm, a signal
that decreases in intensity with the an increase in polymerization time. The two broad
resonances associated with the polymer backbone appear between δ = 5.8
ppm and δ = 6.2 ppm, wherein an increase in intensity is associated with
an increase in polymerization time. A simple ratio of the integrals associated
with these resonances allows the conversion to be
determined at any given time (specifically at greater than 95% for the aromatic
adduct after 6 h), as well as the generation of the kinetic conversion versus
time profiles.
Table 3 shows examples of such profiles and several features that are
worth highlighting. First, the capability of preparing polymers with a high
average DP is desirable for demonstrating the validity of the dendron monomer
approach detailed herein. The dual functional dendron
monomers are susceptible to homopolymerization with Grubbs’ I and III
initiators, albeit with relatively low DP. Then, we evaluated the capability to prepare homopolymers with significantly high
DP. For this series of experiments, we focused exclusively on the 7-oxa-norbornene derivative of the Grubbs’ III initiator with
polymerizations performed in different environments. Table 2 presents
a summary of the theoretical targeted Mn values, together
with the target DP values at quantitative conversion and the SEC-measured number-
and weight-average molecular weights and PDI.
Fluorinated derivative (Entries 1–3) and
ester-containing chain derivative (Entries 4–6) could be
polymerized for quantitative conversion with Grubbs’ I catalyst
within ca 6 h to generate a homopolymer with the SEC measurement. The
reason is that they are all straight chain structures with small steric
hindrance. Hence, the polymerization conditions are considerably easier than
those of other derivatives. By contrast, the use of
Grubbs’ I initiator with trimethoxysilane derivatives (Entries 7–9) resulted in an incomplete conversion with the
siloxy derivative reaching a maximum measured conversion of ca 50%. However,
under these circumstances, near-quantitative conversion was achieved by
substituting the Grubbs’ I species with the Grubbs’ third-generation
initiator, namely, RuCl2(3-BrPy)2 (ImMesH2)CHPh
(Grubbs’ III; data are shown for the trimethoxysilane, aromatic, POSS, and the sugar derivatives) (Entries 10–24). Consistent with previous reports on the enhanced catalytic efficiency of Grubbs’ III species over Grubbs’ I
or II species,28,29 the
polymerizations were rapid with the aromatic derivative and the POSS derivative
reached >95% conversion within ca 6 h
(Entries 13–21).
The steric hindrance effect already became apparent. Special structures, such as sugar structure, and aromatic nucleus, etc. caused a low reactivity. In the
polymerization of bis siloxy derivative, solvents must be
changed from CH2Cl2 to THF to facilitate high
conversions. In addition, the reaction temperature must be raised from
room temperature to 50 oC. The polymerization
time was shortened by 3 h (Entries 7–12), and the polymer
solubility in the organic solvent after sedimentation treatment was poor. After
SEC testing, the test molecular weight greatly
differed from the theoretical molecular weight because the derivative end of silicon oxygen bonds was hydrolytically broken in air
to form a crosslinked network structure.30 The obtained
polymer had certain tensile and thermal stability and a certain degree of ductility, which provided a broad prospect for the
application of such derivatives in materials.31
Aromatic derivatives had a wide value of PDI under the same conditions, but they had suitable values in
toluene at room temperature. Then, the polymerization of
bis aromatic derivatives was more controlled in toluene than in dichloromethane
at room temperature (Entries 16–18). Furthermore, the
reaction time was shortened by 3 h. The performance of difunctional monomer with hydroxyl was unsatisfactory in
THF at an elevated temperature
(Entries 25–27). In general, the characteristics of the end group have an indispensable
influence on the reaction environment (catalyst, solvent and temperature).
The terminal group is POSS. With the growth of the linker group
(alkanolamine), the molecular weight shows a significant increase, but the
molecular weight distribution widens, as presented in Figure 4(A). The longer
the space linker length of the dendritic polymer with the same end
group, the wider the molecular weight distribution. Hence, derivatives with the [Ru] catalyst had a broad molecular weight
distribution, as shown in Table 3. Considering the effect of
steric hindrance on the polymerization of 7-oxa-norbornene ROMP from Figure
4(B), when the linking group is ethanolamine, the greater the steric hindrance
of the terminal group, the greater the PDI. The dendronized
polymer with a chain end had a narrow molecular weight distribution (Entries 1,
4, and 10). A large fluorine content could cause size and phase separation. The molecular
weight distribution of the end straight chain structure is superior to that of the end aromatic or end cage groups (Entries 3, 9,
and 21).
As shown in Table 4 and Figure 5(A), the
reaction time was prolonged with the increase in molecular weight (Entries 1–3).
Moreover, different linker lengths led to a large difference in the
conversion rate of monomers and affected the molecular weight distribution. The
monomer molecular chain length increased and the polydispersity enlarged. When
THF was used as solvent at a heating condition of 50 oC, the molecular
weight distribution was broad (>1.5) for aromatic and ester chain derivatives.
Subsequently, THF was replaced with toluene under various heating conditions (Entries 4–9). The
reaction time was considerably shortened, the conversion was almost
complete, and the molecular weight distribution was within
an acceptable range.
The reaction site of the
tetrafunctional derivative was more than that of the difunctional derivative. After
Michael addition reaction, the products of different functional derivatives were produced, and the polarities were similar, resulting in a low
separation yield. In a series of addition reactions, the isolated yield of
aromatic derivatives was the highest (>70%). However, other thiol compounds, such as
methyl 3-mercaptopropionate and 1H,1H,2H,2H-perfluorodecanethiol, were
challenging for separation with low yields. Through computer
simulation, we found that the steric hindrance energy of different adducts
(one-, two-, and three-adduct products) varied greatly. The reason may be the
low yield of the target product.32 The benzyl group was in a planar configuration,
whereas the other two were in a chain structure, which was prone to torsional winding. The homopolymerization of this
tetrafunctional species proved to be challenging. As the tetrafunctional molecular
weight was considerably greater than the difunctional molecular weight,
for ROMP homopolymerization, the theoretical molecular weight set to 30000
required a complete 12 h conversion in THF 50 oC
with Grubbs’ III catalyst, but PDI >2.5. Furthermore, after replacing the solvent with
toluene, in 50 oC heating for 3 h, the conversion was complete
and an acceptable PDI was obtained. The effect of
chain length was consistent with the difunctional derivatives. As the chain length increased, the obtained molecular
weight distribution widened. However, for a high molecular weight, any improved
reaction conditions were not found.

|
Figure 1 1H NMR spectrum, recorded in CDCl3, of functional exo-7-oxanorbornene monomers G1’’ and G2’’. |

|
Figure 2 1H NMR spectrum, recorded in CDCl3, of the bis siloxane functional dendron monomer with several key peak assignments. |

|
Figure 3 (A) Representative monomer catalyzed by Grubbs catalyst to obtain the dendronized polymers in DCM; (B) conversion versus time profiles generated from the NMR data for the homopolymerization of the benzyl functional dendron monomers, also highlighting the effect of the [Ru] initiator. |

|
Figure 4 SEC traces for (A) the products from the homopolymerization of POSS derivatives with various linker lengths (middle, linker length n = 2; right, linker length n = 3; left, linker length n = 6); (B) the products from the linker length n = 2 and homopolymerization of various terminal groups (left, 1H,1H,2H,2H-perfluorodecanethiol; middle, benzyl mercaptan; and right, methyl 3-mercaptopropionate). |

|
Figure 5 (A) Conversion of difunctional fluorinated derivatives over time under theoretical molecular weight = 30000; (B) SEC traces for the product from the homopolymerization of the tetrafunctional–aromatic derivatives with Grubbs’ III initiator in Tol at 50 oC. Left: linker length = 2. Right: linker length = 6. |
|
Table 3 Summary of Reaction Conditions, Polymerization Time, Measured Average Molecular Weights and Dispersity Values for the Homopolymerization of Monomers Derived from 2-ene with the [Ru] Catalyst (Theoretical Average Molecular Weights = 10 kDa) |

aDetermined using NMR. bDetermined using SEC. |
|
Table 4 Summary of Reaction Conditions, Polymerization Time, Conversion Rate, Measured Average Molecular Weights, and Dispersity Values for the Homopolymerization of Monomers Derived from Difunctional Derivatives with Grubbs’ III Catalyst (Theoretical Average Molecular Weights = 30 kDa) |

aDetermined using NMR. bDetermined using SEC. |
We reported the synthesis of two types of multifunctional dendron
acrylic exo-7-oxanorbornene monomers, namely, difunctional and
tetrafunctional derivatives. 2-ene and 4-ene served as precursors to
thioether-based functional dendron monomers via straightforward nucleophilic
thiol–Michael conjugation between acrylic groups and various
functional thiols, such as siloxane and aromatic thiols. [Ru] kinetics was
demonstrated by NMR, and the monomer was completely converted to a
material with a generally narrow polydispersity under appropriate
conditions. The synthesis and homopolymerization of aromatic derivatives derived from 4-ene proved to be challenging, but such derivatives were
successfully polymerized with Grubbs’ III initiator in toluene at elevated temperature.
The end group affected the functional dendronized
polymer. The shorter the space linker of the dendronized exo-7-oxanorbornene
macromonomer, the narrow molecular weight distributions of the obtained dendronized
polymers.
- 1. K. Liu, Z. Xu, and M. Yin, Prog. Polym. Sci., 46, 25 (2015).
-
- 2. R. H. E. Hudson and M. J. Damha, J. Am. Chem. Soc., 115, 2119 (1993).
-
- 3. L. Song, Q. Jiang, Z.-G. Wang, and B. Ding, ChemNanoMat., 3, 713 (2017).
-
- 4. Y. Shi, W. Zhu, and Y. Chen, Macromolecules, 46, 2391 (2013).
-
- 5. A. Carlmark, C. Hawker, A. Hult, and M. Malkoch, Chem. Soc. Rev., 38, 352 (2009).
-
- 6. N. V. Tsarevsky, S. R. Woodruff, and P. J. Wisian-Neilson, J. Chem. Ed., 93, 1452 (2016).
-
- 7. H. Lee, Polym. Korea, 39, 165 (2015).
-
- 8. D. Sprouse and T. M. Reineke, Biomacromolecules, 15, 2616 (2014).
-
- 9. K. O. Kim, S. Shin, J. Kim, and T.-L. Choi, Macromolecules, 47, 1351 (2014).
-
- 10. A. A. Antonov, N. V. Semikolenova, V. A. Zakharov, W. Zhang, Y. Wang, W.-H. Sun, E. P. Talsi, and K. P. Bryliakov, Organometallics, 31, 1143 (2012).
-
- 11. M. Liu, B. H. Tan, R. P. Burford, and A. B. Lowe, Polym. Chem., 4, 3300 (2013).
-
- 12. L. Zhu, T. J. Zimudzi, N. Li, J. Pan, B. Lin, and M. A. Hickner, Polym. Chem., 7, 2464 (2016).
-
- 13. A. Dondoni and A. Marra, Chem. Soc. Rev., 41, 573 (2012).
-
- 14. M. Liu, J. van Hensbergen, R. P. Burford, and A. B. Lowe, Polym. Chem., 3, 1647 (2012).
-
- 15. J. A. van Hensbergen, M. Liu, R. P. Burford, and A. B. Lowe, J. Mater. Chem. C, 35, 693 (2015).
-
- 16. K. Matyjaszewski, Macromolecules, 45, 4015 (2012).
-
- 17. W. D. Mulhearn and R. A. Register, ACS Macro Lett., 6, 112 (2017).
-
- 18. C. E. Wang, P. S. Stayton, S. H. Pun, and A. J. Convertine, J. Control. Release, 219, 345 (2015).
-
- 19. W. Wieczorek, A. Zalewska, D. Raducha, Z. Florjańczyk, J. R. Stevens, A. Ferry, and P. Jacobsson, Macromolecules, 29, 143 (1996).
-
- 20. V. Percec and D. Schlueter, Macromolecules, 30, 5783 (1997).
-
- 21. S. Rajaram, T.-L. Choi, M. Rolandi, and J. M. J. Fréchet, J. Am. Chem. Soc., 129, 9619 (2007).
-
- 22. K. O. Kim and T.-L. Choi, ACS Macro Lett., 1, 445 (2012).
-
- 23. H. Jung, T. P. Carberry, and M. Weck, Macromolecules, 44, 9075 (2011).
-
- 24. M. Liu, R. P. Burford, and A. B. Lowe, Polym. Int., 63, 1174 (2014).
-
- 25. J. Yamuna, T. Siva, S. S. S. Kumari, and S. Sathiyanarayanan, RSC Adv., 6, 79 (2016).
-
- 26. D. A. Rankin, S. J. P'Pool, H.-J. Schanz, and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 45, 2113 (2007).
-
- 27. M. A. Tallon, Y. Rogan, B. Marie, R. Clark, O. M. Musa, and E. Khosravi, J. Polym. Sci., Part A: Polym. Chem., 52, 2477 (2014).
-
- 28. T.-L. Choi and R. H. Grubbs, Angew. Chem. Int. Ed., 42, 1743 (2003).
-
- 29. G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 110, 1746 (2010).
-
- 30. F. Mangolini, J. Hilbert, J. B. McClimon, J. R. Lukes, and R. W. Carpick, Langmuir, 34, 2989 (2018).
-
- 31. T. Ha, I. Hwang, Y. Bang, K. Kim, J. Kim, S. Kim, and K. Kim, Polym. Korea, 40, 992 (2016).
-
- 32. Z. Zhao, S. Chen, J. W. Y. Lam, C. K. W. Jim, C. Y. K. Chan, Z. Wang, P. Lu, C. Deng, H. S. Kwok, Y. Ma, and B. Z. Tang, J. Phys. Chem. C, 114, 7963 (2010).
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2019; 43(6): 807-815
Published online Nov 25, 2019
- 10.7317/pk.2019.43.6.807
- Received on Jan 11, 2019
- Revised on May 2, 2019
- Accepted on Sep 16, 2019
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Tong Wu, Meina Liu
-
School of Chemical and Environmental Engineering, Shanghai Institute of Technology
- E-mail: 15395472710@163.com, meina.liu@sit.edu.cn
- ORCID:
0000-0002-1736-8472, 0000-0002-9928-2639
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr