






- Preparation and Adhesive Properties of Complex Gels from Polyaspartamides and Tannic Acid
Huong Lan Nguyen Ngoc, Jong Ryoul Moon, and Ji-Heung Kim†

School of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Korea
- 폴리아스팔트아미드와 탄닌산의 컴플렉스 하이드로젤 제조와 그 접착물성
훵란뉜녹 · 문종렬 · 김지흥†
성균관대학교 화학공학과
Several polyaspartamide copolymers were prepared from polysuccinimide, the thermal polycondensation product of aspartic acid, via a successive ring-opening reaction using ethanolamine and other amine nucleophiles. 1H NMR and FTIR spectroscopy were used to identify the chemical structure of the polyaspartamide copolymers. A complex gel was formed upon adding tannic acid (TA) as both a physical cross-linker and adhesive enhancer to the polymer solution at different compositions in an aqueous system. The complexation behavior was investigated and the adhesive properties of the resulting complex gels were measured and compared each other. The thermal properties of the complex gels were also investigated using thermogravimetric analysis. Strong multiple hydrogen-bonding interactions between TA and the polymer chain are considered to be responsible for the formation of these biocompatible complex gels, which have potential use as novel biocompatible adhesive materials.
몇 가지 다른 폴리아스팔트아미드 유도체를 아스팔트산의 열축합 고분자인 폴리숙신이미드의 에탄올아민과 또 다른 아민 화합물을 사용한 연속적인 개환 아미노분해 반응으로부터 제조하였다. 합성한 폴리아스팔트아미드 고분자의 화학적 구조를 NMR과 FTIR 분광기를 사용하여 확인하였다. 물리적 가교제 및 접착물성 증진제로서 탄닌산과 위에서 합성한 고분자를 수용액 상에서 여러 다른 중량비로 혼합함으로써 두 성분간의 컴플렉스 젤을 제조하였다. 이들 컴플렉스 젤 형성 거동과 제조된 젤 시료의 서로 다른 기질과 조건에 대한 접착 물성을 비교 검토하였다. 한편, 열중량분석과 FTIR을 이용하여 재료의 열분해 특성과 두 성분간의 분자간 상호작용을 고찰하였다. 성분 간 강한 수소결합에 기인한 이들 생체적합성 폴리아미노산 고분자와 탄닌산의 컴플렉스 젤은 신규 생체의료용 및 산업용 접착소재로서 응용 가능할 것으로 기대된다.
Several polyaspartamide derivatives with hydroxy, imidazole and primary amine pendants were synthesized and used for complex gel preparation with tannic acid (TA). All the complex gels showed strong adhesive properties on various substrates, where the PHEAEDA/TA complex exhibited the best adhesive strength. These hydrogen-bonding complex polymer gels containing TA have great potential as a biocompatible adhesive and antibacterial soft material.
Keywords: tannic acid, complex gel, polyaspartamide, adhesive properties, hydrogen bonding
This work was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea, funded by the Ministry of Education, Science and Technology (NRF-2016R1D1A1A 09918727).
Hydrogels have network structures that can swell and absorb a large amount of water while maintaining their structure.1 A diversity of hydrogels with a variety of properties have been developed, including exceptional mechanical strength with stimuli-responsiveness, antimicrobial, antibacterial, liquid-gel transition, self-healing, or shape memory effects.2-9 These hydrogels are promising candidates for application in fields such as environmental protection,10 cartilage repair, drug delivery, chemo- and bio-mechanical sensors, and microfluidic devices with hydrogel operated valves and actuators.11-19 Moreover, adhesive properties have an important role in the human body20,21 and have attracted a lot of attention in fields such as wound dressings22 and 3D printing for tissue.23 Using covalent bonding or non-covalent interactions such as hydrogen bonding, π-π interactions, metal-ligand coordination bonding, and guest–host interactions, the corresponding gels can be prepared from a variety of hydrophilic natural and synthetic polymers. Recently, significant interest and research has been focused on the development of smart and functional soft hydrogel materials.
Tannic acid (TA), a specific form of tannin, is a naturally occurring polyphenolic compound found in plants that contains a central glucose unit (carbohydrate core) in which all the hydroxyl groups are esterified with phenols (gallic acid).24 Its weak acidity (pKa ~6) is due to the numerous phenol groups. TA is known for its attractive biological properties, such as antioxidant, anti-bacterial, anti-enzymatic, and astringent activity.25,26 TA is used to treat skin ulcers, wounds, and toothache; its multiple phenolic groups can interact with biological macromolecules. It is, therefore, expected that the addition of TA to synthetic polymers will synergistically enhance their biocompatibility. Due to the presence of the many functional groups in its chemical structure, TA can develop a stable network structure via hydrogen bond interactions, which has been considered as an effective strategy to prepare adhesive materials. Collins et al. added TA to form a boronate ester dynamic covalent cross-linked network within an oxime hydrogel, which was found to increase the hydrogel’s strength.27 Sahiner et al. demonstrated a bulk poly(tannic acid) hydrogel prepared by cross-linking TA molecules with an epoxy cross-linker, which showed good swelling, moisture content behavior, and antimicrobial and antioxidant properties.28 A catechol-based hydrogel has been designed by Andersen et al. that allowed the degree of oxidative covalent cross-linking to be controlled.29 Double cross-linked hydrogels with tunable stiffness were constructed upon adding TA, an oxidizable catechol analogue, to an oxidation-resistant hydrogel held together by coordination of the dihydroxy functionality of 1-(2′-carboxyethyl)-2-methyl-3-hydroxy-4-pyridinone to trivalent metal ions. By varying the amount of TA, the hydrogel stiffness can be customized for a given application, while retaining the self-healing capabilities of the hydrogel’s coordination chemical components. Besides, Guo et al. reported a family of bioadhesives derived from addition of TA to gelatin under oxidizing conditions and cross-linked using silver nitrate.30 The oxidized polyphenol groups of TA enable wet tissue adhesion through catecholamine-like chemistry, while both TA and the silver nanoparticles reduced from silver nitrate provide antimicrobial sources within the polymeric network. Moreover, TA has been introduced to polymer networks (PVA or PAAm) and cross-linked hydrogels via hydrogen bonding, forming double-cross-linked hydrogels (DC).31 The co-operation between the double cross-linked hydrogels results in DC hydrogels that are non-swellable and exhibit strong mechanical strength, large elongation, excellent toughness, good self-recovery, and self-healing abilities in either their as-prepared or swelling equilibrium states. Besides, the chemical nature of TA endows these hydrogels with high adhesive properties to a range of diverse substrates. A poly(N-vinyl pyrrolidone)-TA (PNVP-TA) complex gel studied by Nam et al. shows high adhesive performance on various substrates when compared with neat PNVP polymer.32 In relation to polyphenolic compounds, research on the metal-polyphenol coordination networks has attracted a great attention and recently reviewed.33
Poly(aspartic acid) (PASP), a synthetic poly(amino acid), holds promise as a water-soluble, biodegradable, and biocompatible polymer.34,35 The precursor polymer of PASP, poly (2,5-dioxo-1,3-pyrrolidinediyl), also known as polysuccinimide (PSI), is prepared from aspartic acid, a natural amino acid. Polyaspartamides (PolyAspAm)s include a wide range of PASP amide derivatives that can be prepared using either an aminolysis reaction with PSI or a secondary reaction of the carboxylic pendant groups in PASP. Various aminolysis reactions can then convert PSI into more useful functional polymers or cross-linked gel materials.36-41
Many of these polymers show physicochemical features appropriate for the evolution of biological materials in the form of polymer prodrugs, hydrogels, or nanomaterials. Consequently, stimuli responsive polymers and hydrogel materials have received a great deal of attention in recent years and show substantial promise in the fields of cancer drug delivery and regenerative medicine. To date, a wide range of functional polymers and gels based on polyaspartamides have been investigated toward identifying novel and promising biomaterials.
In recent years, our group has been interested in metal-containing hydrogels and their adhesive properties. The modification of polyaspartamide with imidazole-containing functional groups is also of significant interest due to the strong binding of d-metal ions to imidazole. We have published results on 2-aminoethylimidazole(histamine)-conjugated polyaspartamide and its Cu(II)-coordinated soft gel, which show high adhesive properties on glass and plastic substrates.42 The resulting gel also exhibits reversible self-healing behavior and good antibacterial activity. Tran et al. published results on 3-aminopropyl imidazole(API)-conjugated polyaspartamide and its Ni(II)-coordinated gel, which not only show strong adhesive properties, but also efficient self-healing and rheological behavior.43 In this work, we prepared PHEA, as a basic and reference polyaspartamide, and two polyaspartamide derivatives containing imidazole and alkylamine pendants in order to investigate their complexation behavior with TA. Gelation behavior between TA and the polyaspartamide copolymers in an aqueous solution is observed and the characterization of the resulting gels and their adhesive properties are presented. These simple and non-toxic complex gels have potential use as biocompatible antibacterial adhesive materials.
Materials. L-Aspartic acid (98%), o-phosphoric acid (≥99%), ethanolamine (EA, 99%), 1-(3-aminopropyl)imidazole (API, ≥97%), ethylenediamine (EDA, 99%), tannic acid (TA, ACS reagent), and dimethyl sulfoxide (DMSO, ≥99.7%) were obtained from Sigma-Aldrich Chemical Co. (South Korea). All other chemicals were of sufficient quality for use without purification.
Polymer Synthesis and Complex Gel Preparation Using TA. Synthesis of PSI: L-Aspartic acid (30 g) and o-phosphoric acid (30 g) were placed into a round-bottom flask and mixed at room temperature. The mixture was stirred under reduced pressure from atmospheric pressure to -74 cmHg between 40-180 ℃ and keep at 180 ℃ for 5 h. The reaction mixture was then cooled to room temperature and the resulting semi-solid product was dissolved in DMF. The product was precipitated using excess ethanol and washed several times with de-ionized (DI) water to remove the remaining phosphoric acid. The resulting product was dried at 60 ℃ under vacuum for 3 d. The molecular weight of the final PSI product was determined to be ~24300 Da (l-PSI) and 43200 Da (h-PSI), as calculated from an empirical equation relating the solution viscosity to the molecular weight.44
Synthesis of PHEA: In a three-neck round-bottom flask, 1 g of PSI (l-PSI or h-PSI) was dissolved in 30 mL of DMSO under a nitrogen atmosphere, followed by the addition of excess ethanolamine (EA) and the resulting mixture was stirred at room temperature for 1 d. The solution was then dialyzed and freeze-dried in vacuo to give l-PHEA and h-PHEA (85-90% yield).
Synthesis of PHEA-API: 1 g of PSI (h-PSI) was dissolved in 30 mL of DMSO in a three-neck round-bottom flask equipped with a nitrogen inlet and outlet system. Then, 1-(3-aminopropyl)imidazole (API, 50 mol% based on succinimide unit) was added to the mixture. The resulting solution was stirred at 50 ℃ for 2 d and an excess amount of EA was then introduced at room temperature to react for 1 d. The resulting solution was dialyzed and freeze-dried to give the solid product (70-75% yield).
Synthesis of PHEA-EDA. In a three-neck round-bottom flask, 1 g of PSI (h-PSI) was added and dissolved in 30 mL of DMSO under a nitrogen atmosphere. EA (50 mol% based on succinimide unit) was added and the resulting solution was stirred at room temperature for 24 h. Subsequently, an excess of ethylenediamine (EDA) was introduced very slowly to the reaction mixture at room temperature and stirred for 12 h. The resulting solution was dialyzed and freeze-dried to give the solid product (yield 75-80%).
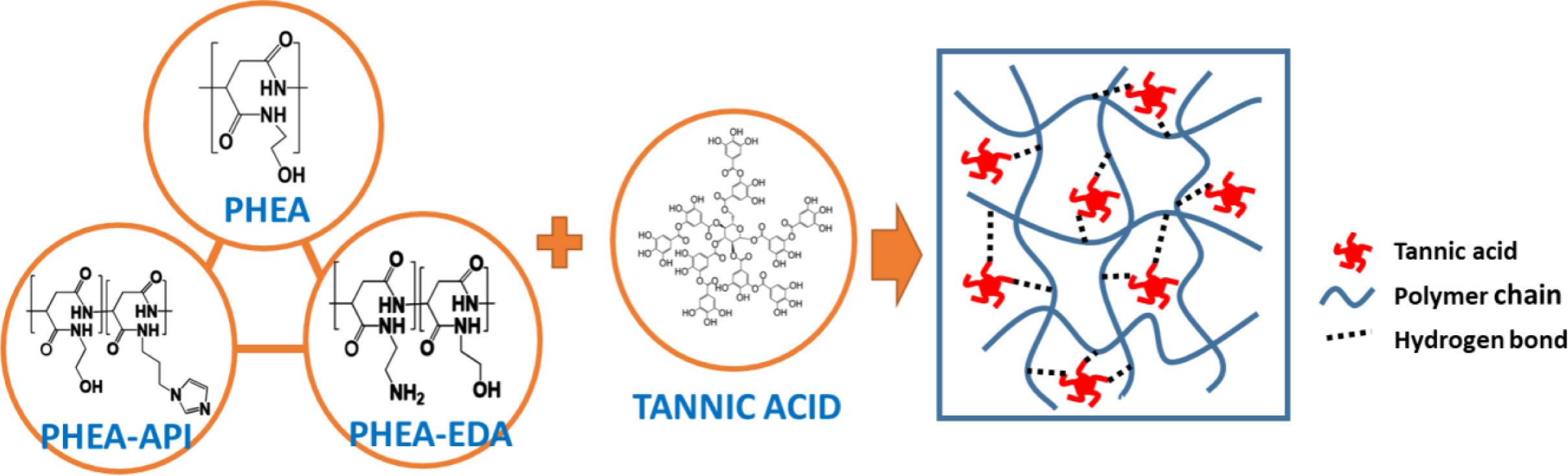
Preparation of PHEA/TA, PHEA-API/TA, and PHEA-EDA/TA Complex Gels: Preparation of the complex hydrogels containing l-PHEA/TA, h-PHEA/TA, PHEA-API/TA, and PHEA-EDA/TA was carried out as follows: The polymer sample and TA were dissolved in DI water at various concentrations using sonication. Thereafter, the TA solution was slowly added dropwise into the polymer solution under stirring. Upon mixing, coacervation or gelation occurred immediately. Next, the coacervate (precipitate) or complex gel formed was washed several times with DI water and freeze-dried. Figure 1 illustrates the chemical structure of polyaspartamides and TA, and the schematic complex gel formation via intermolecular hydrogen bonding interaction.
Characterization and Measurements. Structural Analysis: Proton nuclear magnetic resonance spectroscopy (1H NMR) was recorded on a Bruker AMX-500 spectrometer using D2O as the solvent. A small amount of the dry gels was pelletized upon mixing with KBr powder and used to record the Fourier-transform infrared (FTIR) spectra on an FTIR spectrometer (Perkin Elmer System 200 FTIR).
Thermogravimetric Analysis (TGA): The thermal decomposition behavior of the polymers and gels was evaluated using TGA (TGA7, Perkin Elmer). The samples were analyzed under a nitrogen atmosphere and the temperature increased from room temperature to 600 ℃ at a heating rate of 10 ℃/min.
Water Absorption Capacity: The water absorption (or swelling degree) was measured in DI water at room temperature at certain time intervals. The amount of dry sample was immersed into an aqueous medium for swelling. The excess water was removed from the surface of the sample. The weights of the dry and swollen samples were recorded. The swelling degree was obtained by subtracting the weight of dried sample (Wd) from the weight of swollen sample (Ws), and then dividing this by Wd.
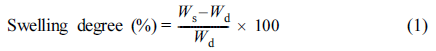
Bulk Adhesion Test. The adhesive strength to different substrates was measured using lap shear strength tests on a universal testing machine (UTM; QC-508E, COMETECH, Taiwan). The test was performed at room temperature using a 500 N load cell at a speed of 15 mm/min. The substrates were glass, PMMA, and aluminum foil (25 mm wide × 75 mm long). The dried gel specimens were mixed with different amounts of DI water and the resulting sticky gels were spread onto a fixed area of one adherend [25 mm (width) × 20 mm (length)]. Then, another adherend was set on the gel-coated adherend and placed for 12 h with compression.
|
Figure 1 Schematic representation of complex formation between polyaspartamide (PHEA, PHEA-API, or PHEA-EDA) and tannic acid (TA). |
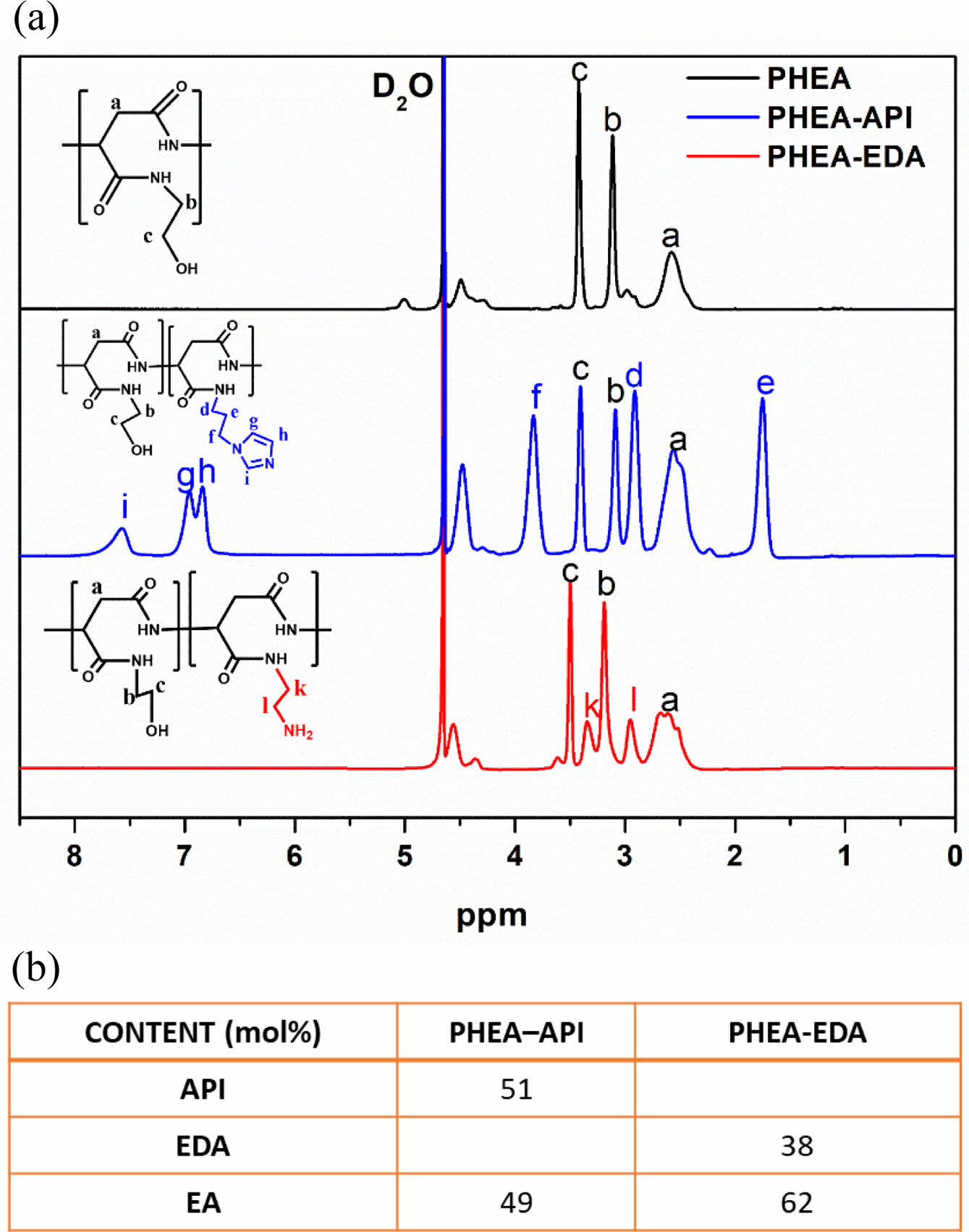
Synthesis and Characterization of PHEA, PHEA-API, and PHEA-EDA. A novel imidazole-modified polyAspAm derivative (PHEA-API) was successfully synthesized from PSI using an aminolysis reaction with imidazole and excess EA. Another polyaspartamide derivative (PHEA-EDA) was prepared using a nucleophilic ring-opening reaction with EDA and EA. Figure 2(a) shows the 1H NMR spectra of PHEA, PHEA-API, and PHEA-EDA. The methylene proton peaks (b and c) that appeared in all the spectra were assigned to the pendent hydroxyethyl groups. Peaks d, e, and f were assigned to the three methylene proton peaks in the API residues. In addition, peaks g, h, and i were assigned to the three heteroaromatic protons on the imidazole ring. Peaks k and l were assigned to methylene protons of the EDA groups. Figure 2(b) shows the compositional data of the PHEA-API and PHEA-EDA copolymers.
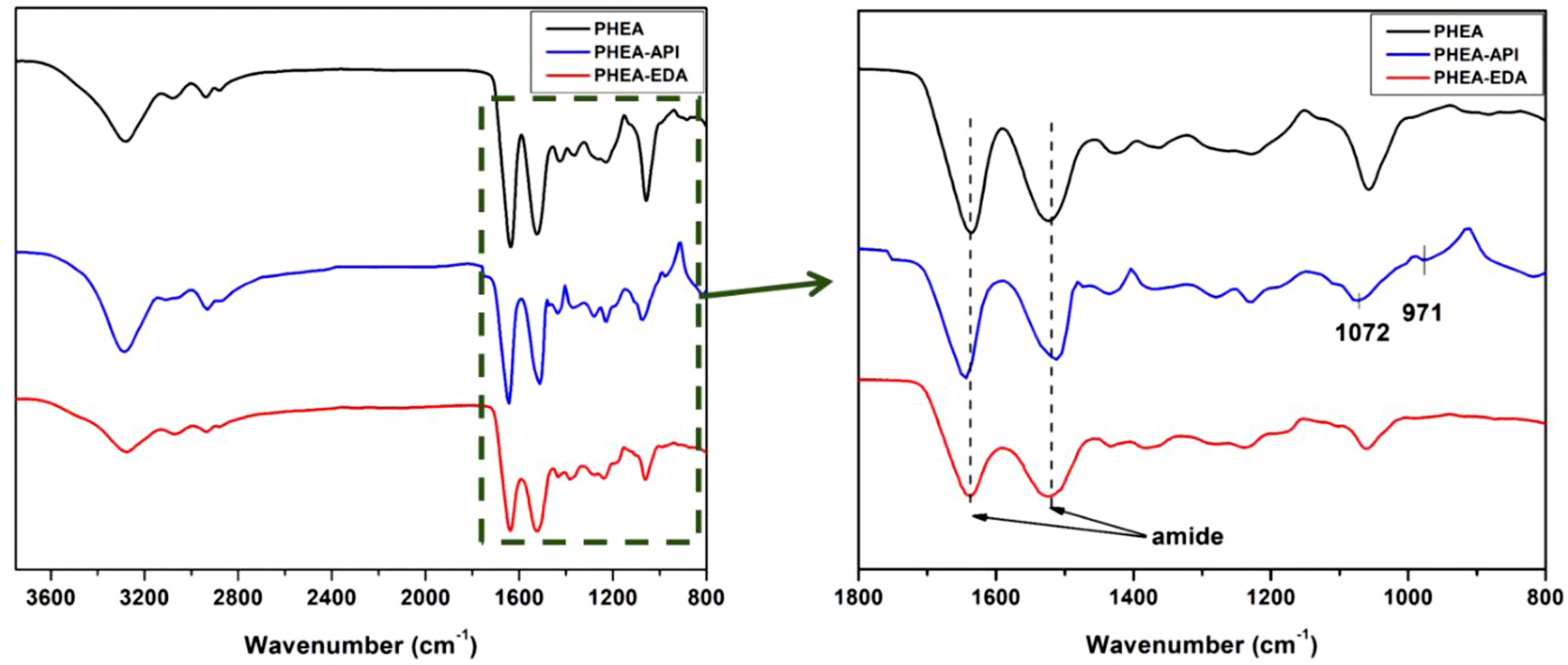
Figure 3 shows the FTIR spectra of PHEA, PHEA-API, and PHEA-EDA. The peaks observed at 1643 and 1511 cm–1 represent the amide I and II stretching vibrations, which were attributed to the ring-opening aminolysis reaction. In the spectra of PHEA-API, the peaks observed at 1072 and 971 cm–1 were assigned to the =C-N stretching and bending vibrations of the imidazole groups. The broad band observed in the range of 3500–3200 cm–1 was attributed to the -OH, -NH, and -NH2 vibrational frequencies of the copolymer, which appear in all the spectra. Since PHEA-EDA contains an amine pendant group, the band corresponding to the -NH stretching and bending vibrations was sharper than that observed in the other polymers.
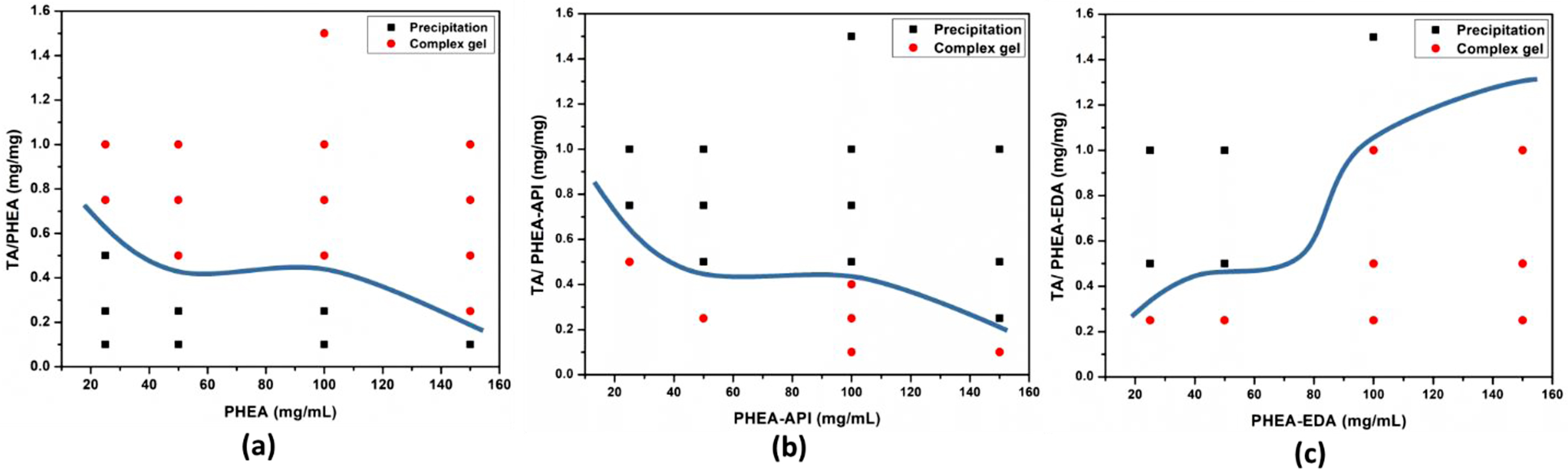
Preparation of PHEA/TA, PHEA-API/TA, and PHEA-EDA/TA Complex Gels. The complex gels were prepared using PHEA, PHEA-API, and PHEA-EDA, in which each polymer was reacted with TA upon mixing at various concentrations. Figure 4 shows photographs of the complex gels or precipitates formed upon mixing the two components in an aqueous solution. Depending on the concentration and relative TA content, the mixed solution formed a precipitate or sticky gel. Figure 5 illustrates the rough phase diagrams of the three different polymer/TA systems depending on the relative TA content and solution concentration of each polymer, i.e., PHEA, PHEA-API, and PHEA-EDA. The mixtures form either a complex gel or precipitate at different compositions. In these diagrams, the boundary between the complex gel and precipitate varies according to the different polymer/TA systems studied. For the PHEA/TA system, complex gel formation is favorable at a high TA content and increasing polymer concentration. At a low TA ratio and low polymer concentration, the mixture tends to form a precipitate. On the other hand, the PHEA-API/TA system shows different behavior, where gelation occurred at a low TA content and over a wide range of polymer concentrations. The PHEA-EDA/TA system demonstrates a similar phase diagram to PHEA-API/TA, but in an opposite way in terms of the TA content and polymer concentration. Complex intermolecular interactions, including hydrogen bonding in the presence of water, among these multi-functional polymers and TA are responsible for the different phase behavior, but a detailed elucidation of this phenomena falls out of the scope of this communication.
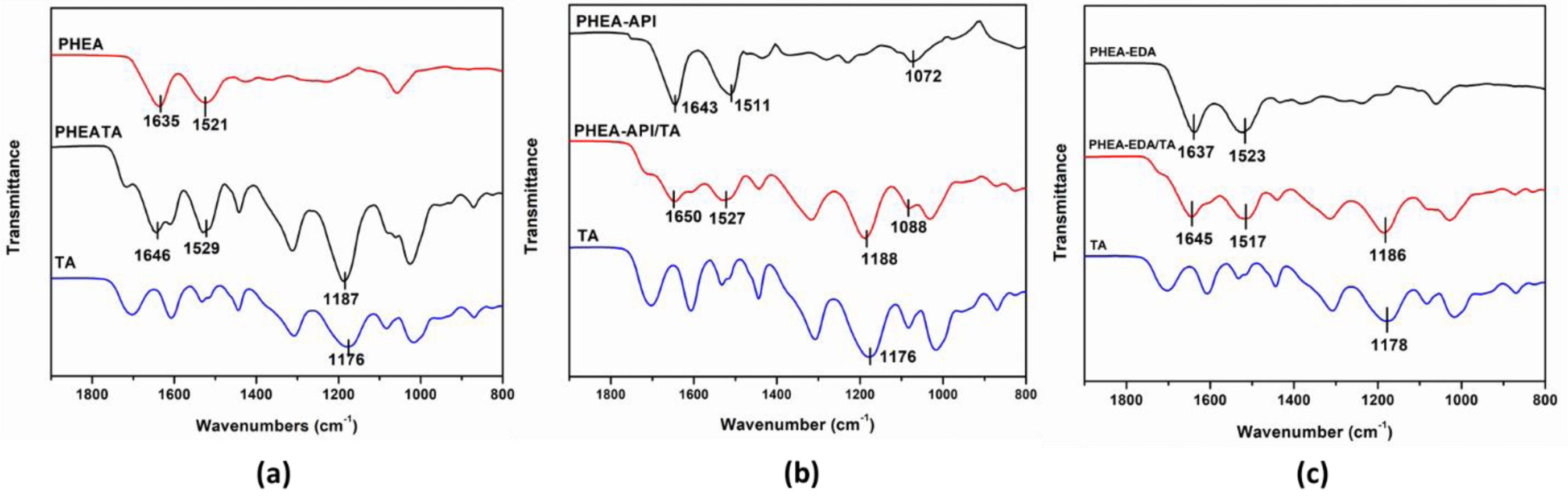
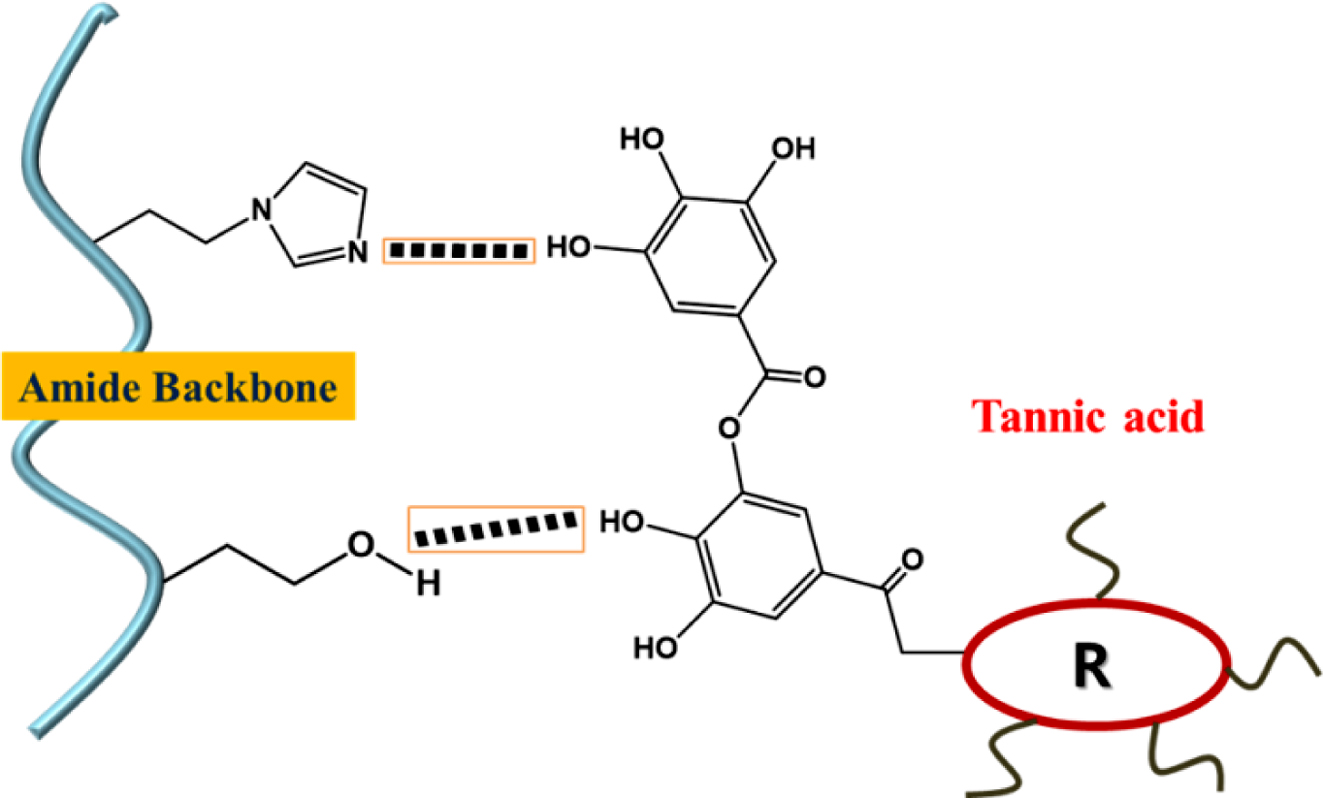
Characterization of PHEA/TA, PHEA-API/TA, and PHEA-EDA/TA Complex Gels. To obtain information on the intermolecular interactions, FTIR spectroscopy was carried out (Figure 6). For TA, the spectrum showed some characteristic bands, i.e., the deformation vibrations of the C-C bonds in the phenolic groups in the region of 1500-1400 cm–1, stretching vibrations of the -C-C aromatic groups at ~1442 cm–1, and characteristic C=O stretching vibrations at 1706 cm–1 and C-O at 1100-1300 cm–1, which are ascribed to the aromatic esters groups in TA. In the spectrum of the PHEA/TA complex, the characteristic absorption bands of the two components were observed. In addition, the original amide I and II bands of PHEA at 1635 and 1521 cm–1 were shifted to 1646 and 1529 cm–1, respectively. In addition, the band at 1176 cm–1 corresponding to the C-O vibrations in TA was shifted to 1187 cm–1, suggesting strong hydrogen bonding interactions were formed between the polymer and TA molecules. In the case of the PHEA-API/TA complex, the amide bands of PHEA-API changed from 1643 and 1511 cm–1 to 1650 and 1527 cm–1, respectively, and the C-O peak of TA shifted from 1176 to 1188 cm–1. In addition, the absorption band of =C-N bond changed from 1072 to 1088 cm–1 due to the hydrogen bonding interactions formed between the phenolic hydroxy and imidazole groups in PHEA-API. For the PHEA-EDA/TA system, the amide bands of the polymer changed from 1637 and 1523 cm–1 to 1645 and 1517 cm–1, respectively. In addition, the C-O band of TA was shifted from 1178 to 1186 cm–1. This FTIR analysis confirms the intermolecular interactions, including hydrogen bonding, between the different polyaspartamides and TA, and a representation of these intermolecular interactions is shown in Figure 7.
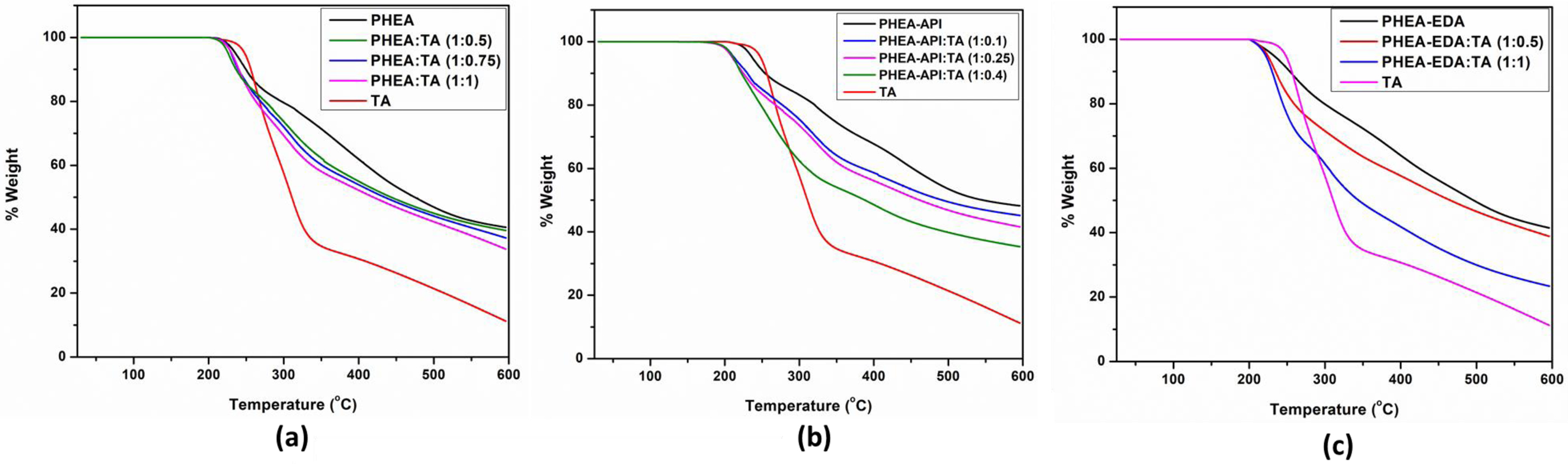
Meanwhile, TGA was performed under a nitrogen atmosphere to investigate the thermal stability of the polymer-TA complexes. As shown in Figure 8(a), the decomposition temperatures of PHEA and TA were 225 and 242 ℃, respectively. As seen from the curves, the thermal decomposition onset temperature of the PHEA/TA complex was lower when compared its two components, suggesting its reduced thermal stability. In the presence of TA, the thermal stability of the polymer appears to decrease. Two steps of decomposition were observed for the PHEA/TA gel, which were attributed to the individual thermal decomposition steps of the TA and PHEA species in the gel upon increasing the temperature. In the case of the PHEA-API/TA system, the complex gel showed a noticeably lower decomposition onset temperature than those of its components. The decomposition temperatures of PHEA-API and PHEA-API:TA (1:0.4) were observed at ~230 and 200 ℃, respectively, which suggest that the thermal degradation of the polymer was accelerated in the presence of TA. Upon increasing the amount of TA, the weight loss of the samples gradually increased, as shown in Figure 8(b). In the PHEA-EDA/TA system, the decomposition onset temperatures of PHEA-EDA and PHEA-EDA/TA were nearly identical (~220 ℃) (Figure 8(c)). From the residual weight at 600 ℃ obtained from the TGA data, we can roughly estimate the actual TA content within the complex gel. For the PHEA/TA system, the amount of TA was estimated to be 5.7, 12.5, and 24.9 wt% for the samples prepared with feed ratios of 1:0.5, 1:0.75, and 1:1, respectively. Similarly, for the PHEA-API/TA system, the actual TA contents were calculated to be 9.2, 16.5, and 34 wt% at feed ratios of 1:0.1, 1:0.25, and 1:0.4, respectively, and for PHEA-EDA:TA, 10.7 and 60.4 wt% was obtained at feed ratios of 1:0.5 and 1:1, respectively.
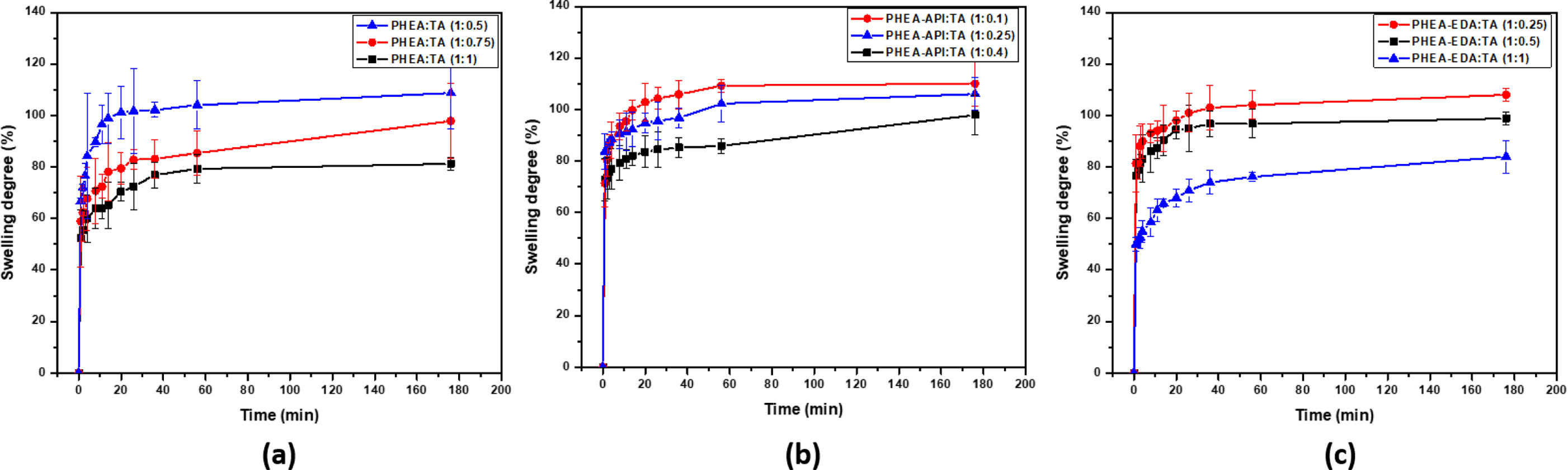
Water Absorption. The swelling curves of the different complex gels prepared using different TA contents were obtained. Figure 9 shows that these complex gels reach their equilibrium swelling in about 1 h and the water absorption values were in the range of 70-110%. Upon increasing the TA content, the water absorption decreased, which was attributed to the effect of increasing the amount of physical cross-linking within the gel network.
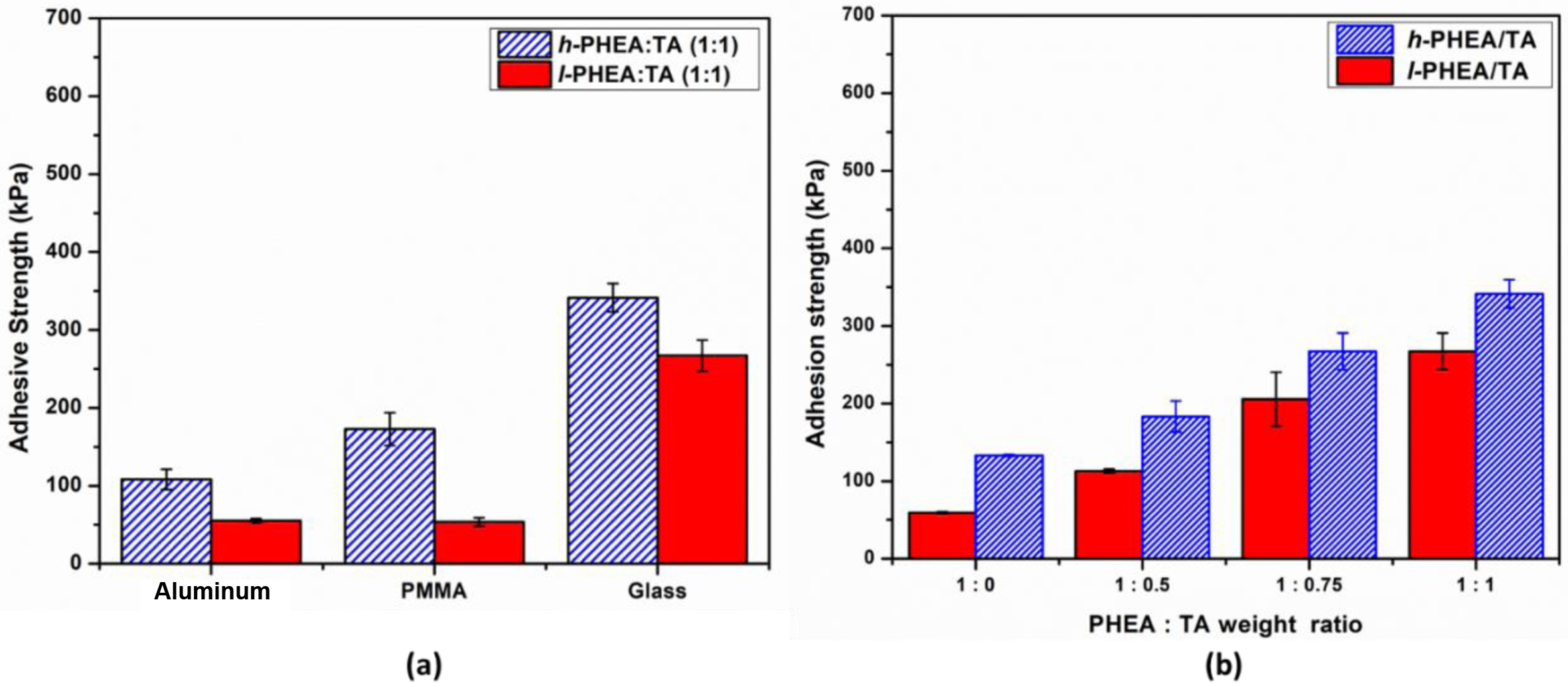
Adhesive Properties of Complex Gels. To measure the adhesive properties of the three complex gel systems: PHEA/TA, PHEA-API/TA, and PHEA-EDA/TA, a single lap shear test was carried out, which was repeated in triplicate for each sample. There are a diverse range of parameters in the complex gel that can affect the adhesive properties, such as the molecular weight of polymer, gel composition, water content, type of adherend, and aging factor. Figure 10(a) describes the adhesive strength of the complex gels prepared from l-PHEA or h-PHEA and TA. High molecular weight polymers afford higher adhesive strength than low molecular weight polymers due to efficient chain entanglement. For the same composition of PHEA:TA (1:1), the adhesive strength values of h-PHEA/TA (342±18, 173±21, and 109±13 kPa) were higher than l-PHEA/TA (269±20, 53±6, and 55±2 kPa) observed for the glass, PMMA, and aluminum substrates, respectively. The dependence of the adhesive strength on a glass substrate on the PHEA/TA ratio is shown in Figure 10(b). The adhesive strength of the single polymer without TA was much lower than that of the TA complex gels, which clearly indicates the important role of TA in the adhesive properties. Upon increasing the TA content in the complex, the adhesive strength increased linearly. In all the different PHEA:TA ratios studied, the h-PHEA/TA complex gel exhibited higher adhesive strength when compared to l-PHEA/TA.
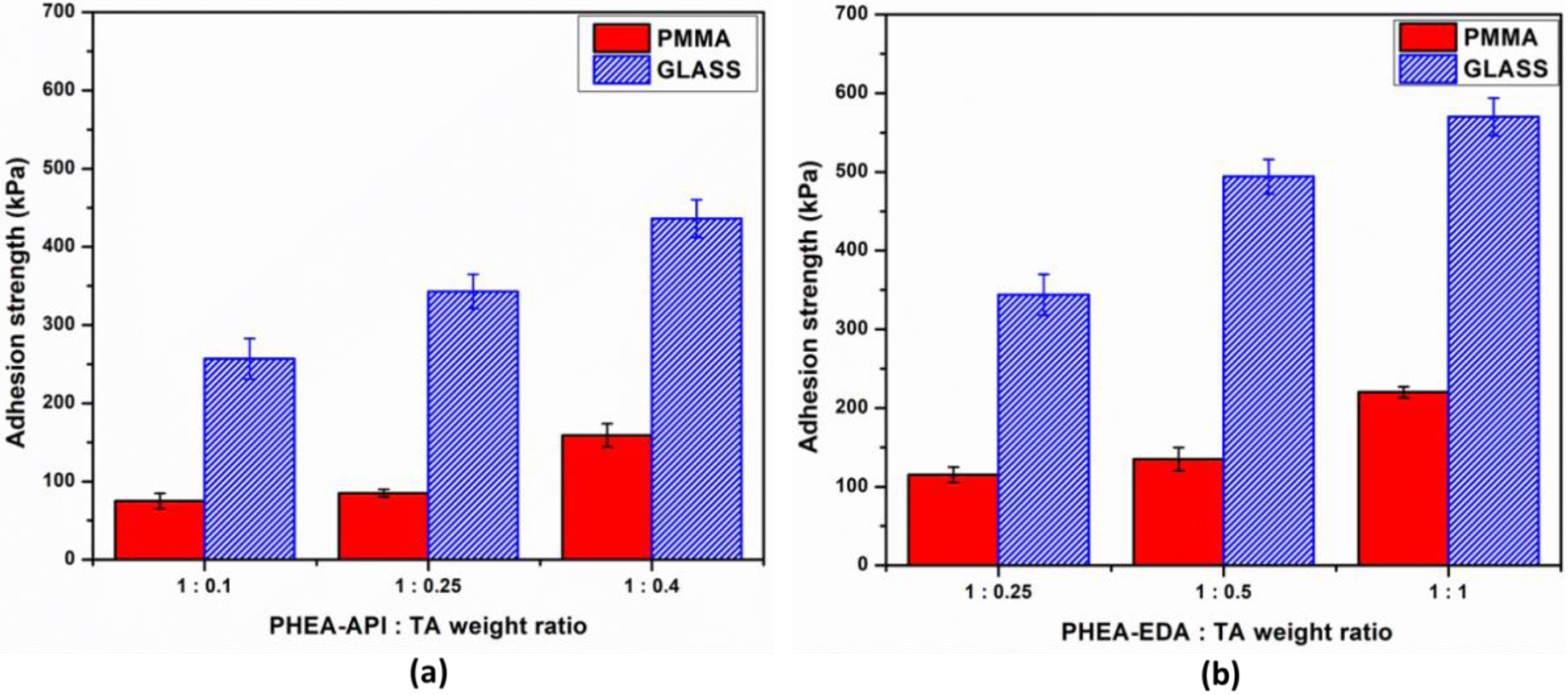
Figure 11 shows the adhesive properties of the PHEA-API/TA and PHEA-EDA/TA gel systems with various composition ratios on glass and PMMA substrates. As can be clearly seen, both complex gels exhibited greater adhesion on glass than PMMA. For example, PHEA-API/TA at a ratio of 1:0.4 has an adhesive strength of 159±15 and 436±24 kPa on the PMMA and glass substrates, respectively. Again, an increasing polymer/TA weight ratio results in the higher adhesive strength of the gel material. The overall adhesive strength on PMMA, one of the common plastics, was observed to be relatively low due to the rather hydrophobic nature of the PMMA surface. However, the adhesive strength on glass, an inorganic silicate, was extraordinarily high, suggesting the very strong interactions formed between the TA containing polymer gel containing imidazole or amine functional groups and the polar ionic silicate surface. When compared to the PHEA-API/TA system, the adhesive strength of the PHEA-EDA/TA gel was about 1.4 times higher.
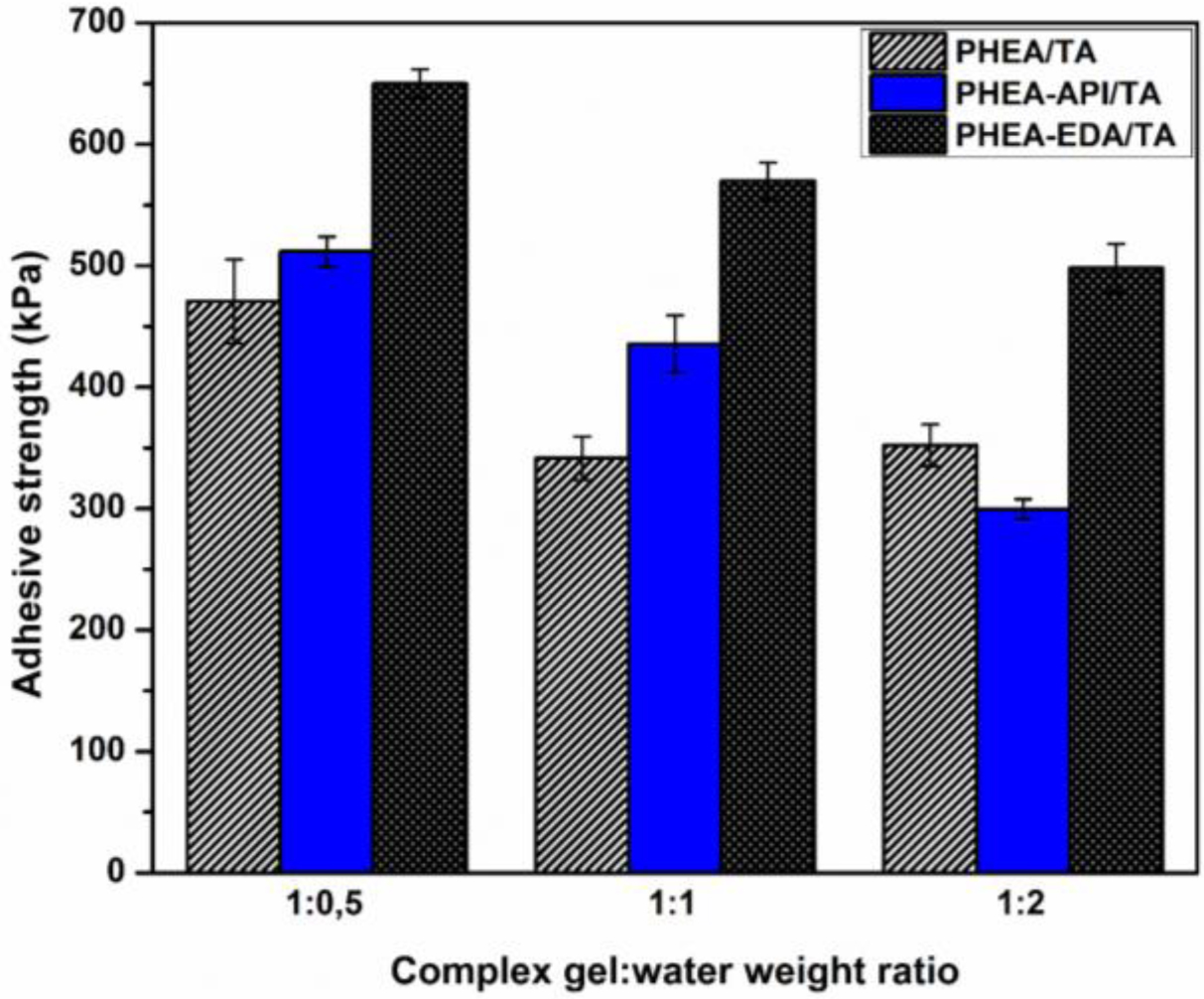
Next, the effect of water in the adhesive mixture was tested to elucidate the influence of residual water on the adhesive performance of the complex gels studied. Figure 12 illustrates the results for the adhesive properties on a glass substrate for the h-PHEA/TA and PHEA-API/TA complex gel systems at various gel:water ratios. At a dry gel:water ratio of 1:0.5, the highest adhesive strength was observed and the apparent adhesive strength decreased as the water content increased. This result strongly suggests that water molecules at the adhesion layer play an important role in the adhesion performance. An excess amount of water should prevent any strong interactions being formed between the adhesive matter and substrate, which consequently reduces the adhesive strength.
To summarize, the adhesive properties of the TA complex gels prepared using the three different polyaspartamides were investigated under ambient conditions. The adhesive strength varied according to the different polymer structures, relative composition with TA, and water content in the adhesive formulation. A high level of adhesive strength was observed on the glass substrate, however, the adhesive strength was much lower on PMMA. Among the three different polyaspartamide derivatives, PHEA-EDA displays the highest adhesive strength of 570±15 and 220±7 kPa on glass and PMMA, respectively. This indicates that the interaction and binding between TA and the amine groups is more efficient compared to imidazole or hydroxy groups, which may be caused by the more basic nature of primary amine along with its strong hydrogen-bonding capability.
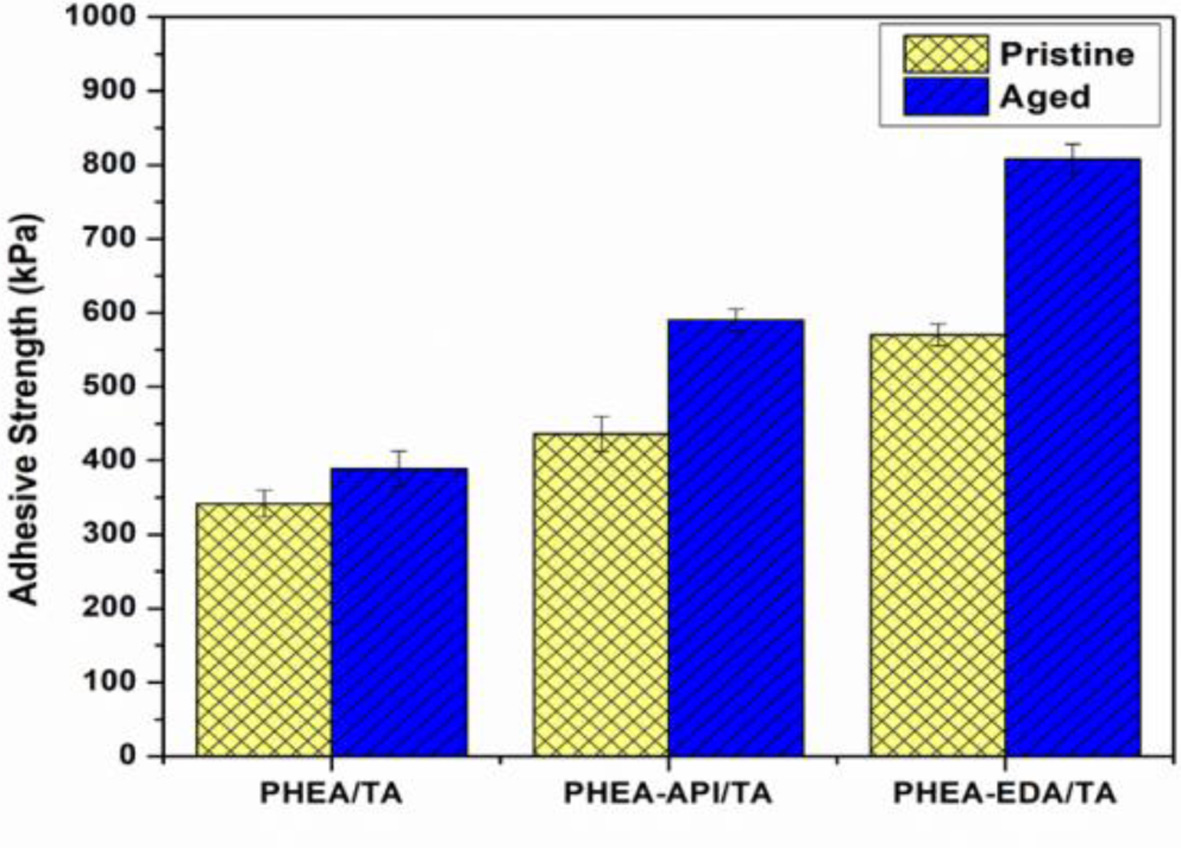
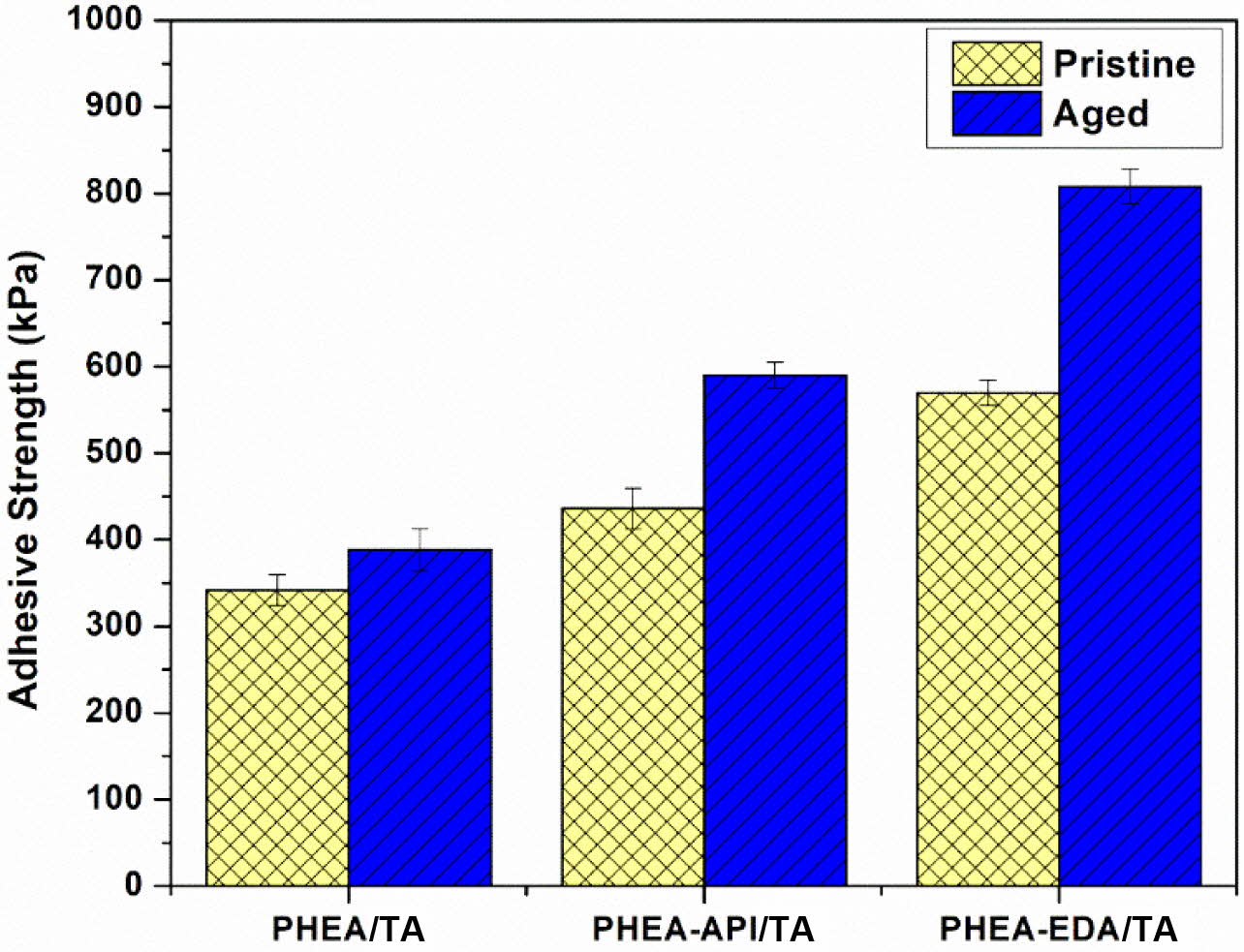
Lastly, the aging effect on the adhesive properties was examined using three representative polyAspAm/TA complex gels, which showed enhanced adhesive performance. Each test sample were prepared on a glass substrate and the test specimens were placed in an oven at 60 ℃ for 2 h before the lap shear test. Figure 13 shows the results and clearly demonstrates that the adhesive strength after aging was significantly improved when compared to the previous data obtained from the untreated samples. In particular, for the PHEA-EDA/TA system, the adhesive strength was increased significantly up to 808±20 kPa when compared to 570±15 kPa found for the original sample without thermal treatment. A more detailed study seems to be necessary to fully understand the aging effect at different conditions. However, the aging process appears to increase the adhesive performance through a more stable consolidation of the adhesive layer upon heating and a possible chemical cross-linking reaction between the component materials.
|
Figure 2 (a) 1H NMR spectra of PHEA, PHEA-API, and PHEA-EDA; (b) The composition of each pendant group in the polyAspAm copolymers. |
|
Figure 3 FTIR spectra of PHEA, PHEA-API, and PHEA-EDA. |

|
Figure 4 Representative photographs of gel or precipitate formation via complexation between polyaspartamide and TA: (a) Precipitation at TA/PHEA = 0.2; (b) complex gel formation at TA/PHEA = 1 in the concentration of 100 mg/mL. |
|
Figure 5 Complexation behavior dependence on TA content and polymer concentration observed for (a) PHEA/TA; (b) PHEA-API/TA; (c) PHEA-EDA/TA systems. |
|
Figure 6 FTIR spectra of TA, polymer, and polymer-TA complex: (a) PHEA/TA; (b) PHEA-API/TA; (c) PHEA-EDA/TA. |
|
Figure 7 Intermolecular hydrogen bonding interactions between polyaspartamide and TA. |
|
Figure 8 TGA curves obtained for (a) PHEA/TA; (b) PHEA-API/TA; (c) PHEA-EDA/TA systems. |
|
Figure 9 Swelling curves obtained for (a) PHEA/TA; (b) PHEA-API/TA; (c) PHEA-EDA/TA gels. |
|
Figure 10 Effect of the molecular weight of PHEA on the adhesive property of PHEA/TA system: The adhesive strength of high or low molecular weight PHEA/TA complex gels toward different substrates (a); the adhesive strength on glass substrate depending on PHEA:TA weight ratio (b). |
|
Figure 11 Adhesive strength of (a) PHEA-API/TA gel; (b) PHEA-EDA/TA gel at different polymer:TA weight ratios. |
|
Figure 12 Effect of water on the adhesive properties: h-PHEA/TA and PHEA-API/TA complex gel on a glass substrate at different gel:water weight ratios. |
|
Figure 13 The aging effect on the adhesive strength of PHEA/TA, PHEA-API/TA, and PHEA-EDA/TA complex gels on a glass substrate (60 oC, 2 h). |
Novel copolymers based on polyaspartamide modified with ethanolamine, 1-(3-aminopropyl)-imidazole, or ethylenediamine were synthesized. An adhesive complex gel was prepared by mixing aqueous TA with polyaspartamide solution under vigorous stirring at different compositions. All the complex gels showed appreciable adhesive properties on various substrates. Among them, the PHEA-EDA/TA complex showed higher adhesive strength than the other systems and its adhesive strength on a glass substrate, in particular, was extremely high. As the water content in the complex gel increased, the adhesive strength decreased due to both the TA and polymers being relatively hydrophilic. From the aging effect at elevated temperature, the adhesive strength values increased for all the systems studied. To conclude, it has been demonstrated that complex gels containing TA and polyaspartamide copolymers possess strong adhesive properties on various material surfaces, suggesting their great potential as biocompatible adhesive materials for many biomaterial applications.
- 1. Y. Qiu and K. Park, Adv. Drug Deliv. Rev., 53, 321 (2001).
-
- 2. X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui, and W. Liu, Adv. Mater., 27, 3566 (2015).
-
- 3. J. Ma, Y. Shen, C. Shen, Y. Wen, and W. Liu, Chem. Eng. J., 248, 98 (2014).
-
- 4. Y. Qin, L. L. Chen, W. Pu, P. Liu, S. X. Liu, Y. Li, X. L. Liu, Z. X. Lu, L. Y. Zheng, and Q. E. Cao, Chem. Commun., 55, 2206 (2019).
-
- 5. M. Li, X. Jiang, D. Wang, Z. Xu, and M. Yang, Colloids Surf., B, 177, 370 (2019).
-
- 6. M. Malmsten, Soft Matter, 7, 8725 (2011).
-
- 7. S. Naficy, H. R. Brown, J. M. Razal, G. M. Spinks, and P. G. Whitten, Aust. J. Chem., 64, 1007 (2011).
-
- 8. R. Messing and A. M. Schmidt, Polym. Chem., 2, 18 (2011).
-
- 9. P. Calvert, Adv. Mater., 21, 743 (2009).
-
- 10. S. S. Cutié, P. B. Smith, D. E. Henton, T. L. Staples, and C. Powell, J. Polym. Sci., Part B: Polym. Phys., 35, 2029 (1997).
-
- 11. V. K. Thakur and M. K. Thakur, Hydrogels: Recent Advances, Springer, Singapore, 2018.
-
- 12. M. A. Hempenius, C. Cirmi, F. L. Savio, J. Song, and G. Vancso, J. Macromol. Rapid Commun., 31, 772 (2010).
-
- 13. G. Y. Huang, L. H. Zhou, Q. C. Zhang, Y. M. Chen, W. Sun, F. Xu, and T. Lu, J. Biofabrication, 3, 012001 (2011).
-
- 14. S. Allazetta, S. Cosson, and M. O. Lutolf, Chem. Commun., 47, 191 (2011).
-
- 15. C. W. Peak, J. J. Wilker, and G. Schmidt, Colloid Polym. Sci., 291, 2031 (2013).
-
- 16. H. J. Schneider, K. Kato, and R. M. Strongin, Sensors, 7, 1578 (2007).
-
- 17. J. Texter, Colloid Polym. Sci., 287, 313 (2009).
-
- 18. D. Kuckling, Colloid Polym. Sci., 287, 881 (2009).
-
- 19. A. Matsumoto, R. Yoshida, and K. Kataoka, Biomacromolecules, 5, 1038 (2004).
-
- 20. C. A. Anderson, A. R. Jones, E. M. Briggs, E. J. Novitsky, D. W. Kuykendall, N. R. Sottos, and S. C. Zimmerman, J. Am. Chem. Soc., 135, 7288 (2013).
-
- 21. B. P. Hung, O. M. Babalola, and L. J. Bonassar, J. Biomed. Mater. Res. A, 101, 3592 (2013).
-
- 22. B. Mirani, E. Pagan, B. Currie, M. A. Siddiqui, R. Hosseinzadeh, P. Mostafalu, Y. S. Zhang, A. Ghahary, and M. Akbari, Adv. Healthc. Mater., 6, 1700718 (2017).
-
- 23. S. Hong, D. Sycks, H. F. Chan, S. Lin, G. P. Lopez, F. Guilak, K. W. Leong, and X. Zhao, Adv. Mater., 27, 4035 (2015).
-
- 24. H. A. Cheng, C. T. Drinnan, N. Pleshko, and O. Z. Fisher, Soft Matter, 11, 7783 (2015).
-
- 25. T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau, and P. B. Messersmith, Angewandte Chemie, 125, 10966 (2013).
-
- 26. S. Vijayalaxmi, S. K. Jayalakshmi, and K. Sreeramulu, J. Food Sci. Technol., 52, 2761 (2015).
-
- 27. J. Collins, M. Nadgorny, Z. Xiao, and L. A. Connal, Macromol. Rapid Commun., 38, 1600760 (2017).
-
- 28. N. Sahiner, S. Sagbas, M. Sahiner, C. Silan, N. Aktas, and M. Turk, Int. J. Biol. Macromol., 82, 150 (2016).
-
- 29. A. Andersen, M. Krogsgaard, and H. Birkedal, Biomacromolecules, 19, 1402 (2018).
-
- 30. J. Guo, W. Sun, J. P. Kim, X. Lu, Q. Li, M. Lin, O. Mrowczynski, E. B. Rizk, J. Cheng, G. Qian, and J. Yang, Acta Biomater., 72, 35 (2018).
-
- 31. H. Fan, J. Wang, and Z. Jin, Macromolecules, 51, 1696 (2018).
-
- 32. H. G. Nam, M. G. Nam, P. J. Yoo, and J. H. Kim, Soft Matter, 15, 785 (2019).
-
- 33. K. Halake, S. Cho, J. Kim, T. Lee, Y. Cho, S. Chi, M. Park, K. Kim, D. Lee, H. Ju, Y. Choi, M. Jang, G. Choe, and J. Lee, Macromol. Res., 26, 93 (2018).
-
- 34. E. Jalalvandi and A. Shavandi, Eur. Polym. J., 109, 43 (2018).
-
- 35. G. Wang, Y. Xiao, H. Xu, P. Hu, W. Liang, L. Xie, and J. Jia, J. Agric. Food Chem., 66, 11244 (2018).
-
- 36. J. R. Moon and J. H. Kim, Polym. Int., 59, 630 (2010).
-
- 37. J. R. Moon, M. W. Kim, D. Kim, J. H. Jeong, and J. H. Kim, Colloid Polym. Sci., 289, 63 (2011).
-
- 38. J. R. Moon, Y. S. Jeon, M. Zrinyi, and J. H. Kim, Polym. Int., 62, 1218 (2013).
-
- 39. N. T. Huynh, Y. S. Jeon, D. Kim, and J. H. Kim, Polymer, 54, 1341 (2013).
-
- 40. G. Giammona, G. Pitarresi, G. C. Carlisi, E. F. Craparo, and D. Mandracchia, J. Drug Deliv. Sci. Technol., 16, 77 (2006).
-
- 41. G. Pitarresi, P. Pierro, F. S. Palumbo, G. Tripodo, and G. Giammona, Biomacromolecules, 7, 1302 (2006).
-
- 42. J. R. Moon, Y. S. Jeon, Y. J. Kim, and J. H. Kim, J. Polym. Res., 26, 12 (2019).
-
- 43. N. B. Tran, J. R. Moon, Y. S. Jeon, J. Kim, and J. H. Kim, Colloid Polym. Sci., 295, 655 (2017).
-
- 44. P. Neri, G. Antoni, F. Benvenuti, F. Cocola, and G. Gazzei, J. Med. Chem., 16, 893 (1973).
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2019; 43(5): 705-715
Published online Sep 25, 2019
- 10.7317/pk.2019.43.5.705
- Received on Apr 23, 2019
- Revised on May 14, 2019
- Accepted on May 26, 2019
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Ji-Heung Kim
-
School of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Korea
- E-mail: kimjh@skku.edu
- ORCID:
0000-0002-7767-6952
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr